
Domesztikált kannabinoid-szintázok egy vad mozaikos kannabisz-pángenomban
Eredeti angol cím: Domesticated cannabinoid synthases amid a wild mosaic cannabis pangenome
Szerzők: Ryan C. Lynch, Lillian K. Padgitt-Cobb, Andrea R. Garfinkel, Brian J. Knaus, Nolan T. Hartwick, Nicholas Allsing, Anthony Aylward, Philip C. Bentz, Sarah B. Carey, Allen Mamerto, Justine K. Kitony, Kelly Colt, Emily R. Murray, Tiffany Duong, Heidi I. Chen, Aaron Trippe, Alex Harkess, Seth Crawford, Kelly Vining és Todd P. Michael
Intézményi hovatartozások: 1 The Plant Molecular and Cellular Biology Laboratory, The Salk Institute for Biological Studies, La Jolla, CA, USA; 2 Oregon CBD, Independence, OR, USA; 3 Department of Horticulture, Oregon State University, Corvallis, OR, USA; 4 HudsonAlpha Institute for Biotechnology, Huntsville, AL, USA; 5 Department of Cell and Developmental Biology, School of Biological Sciences, University of California San Diego, La Jolla, CA, USA; 6 Science and Conservation, San Diego Botanical Garden, Encinitas, CA, USA; 7 Center for Marine Biotechnology and Biomedicine, University of California San Diego, La Jolla, CA, USA
Egyenlő szerzői hozzájárulás: Ryan C. Lynch és Lillian K. Padgitt-Cobb egyenlő mértékben járultak hozzá a munkához.
Kapcsolat: rlynch@colorado.edu; lilliankpc@gmail.com; toddpmichael@gmail.com
Előzménydátumok: érkezett: 2024. május 21.; elfogadva: 2025. április 24.; online megjelent: 2025. május 28.; Nature 643, 1001–1010 (2025), 2025. július 24-i lapszám.
Lynch et al.: a kannabisz génkészletének és evolúciós történetének pángenomikai újraértelmezése
Ez a tanulmány a sorozat legújabb és legnagyobb felbontású állomása. Míg a 2020-as munka a botanikai-rendszertani alakköröket rendezi újra, a 2023-as áttekintés pedig a genomikai taxonómia felől értelmezi a kannabisz vitatott osztályozását, Lynch és munkatársai már pángenomikai szinten vizsgálják a növényt. Ez azt jelenti, hogy nem egyetlen referencia-genom alapján próbálják leírni a kannabiszt, hanem sok különböző genetikai hátterű növény teljesebb génkészleti változatosságát térképezik fel.
A kutatás 181 új és 12 korábban publikált genom felhasználásával épít kannabisz-pángenomot, 144 biológiai mintából, hím és nőivarú növényeket egyaránt bevonva. A cél nem pusztán az, hogy pontosabb genetikai térkép készüljön, hanem hogy láthatóvá váljon a fajon belüli mélyebb szerkezeti változatosság: a populációs mintázatok, a történeti hibridizáció, az ivari kromoszómák evolúciója, a transzponálható elemek szerepe, a strukturális variációk, valamint a kannabinoidszintáz-gének különleges helyzete.
A tanulmány különösen fontos a „Sativa–Indica–Hybrid” kereskedelmi gondolkodás meghaladásában. A pángenomikai adatok alapján a kannabisz nem két vagy három egyszerű piaci kategóriába rendeződik, hanem mozaikos, hibridizált, szerkezetileg változékony és részben még feltáratlan génkészletű növényként jelenik meg. A modern fajták, a rostkenderek, a magas kannabinoidtartalmú hemp-vonalak, a drogtípusú populációk és az ázsiai vad vagy vad jellegű génkészletek nem választhatók szét pusztán címkék alapján.
A tanulmány egyik stratégiai jelentőségű megállapítása, hogy a ma ismert és kutatásokban gyakran használt kannabiszgenomok még nem fedik le a faj teljes természetes változatosságát: a nyugati nemesítési vonalak, modern drogtípusú hibridek, rostkenderek és CBD-domináns fajták csak a kép egy részét adják. A pángenomikai adatok arra utalnak, hogy Ázsiában — különösen a kevéssé mintázott vad, elvadult és tájfajta jellegű állományokban — olyan mélyebb genetikai ágak és génváltozatok maradhattak fenn, amelyek nem illeszkednek tisztán a mai kereskedelmi „Sativa–Indica–Hybrid” logikába.
Hivatkozás: Lynch RC, Padgitt-Cobb LK, Garfinkel AR és munkatársai (2025) Domesticated cannabinoid synthases amid a wild mosaic cannabis pangenome. Nature 643: 1001–1010. DOI: 10.1038/s41586-025-09065-0
Eredeti cikk: https://www.nature.com/articles/s41586-025-09065-0
Szerzői jog és licenc: A cikk nyílt hozzáférésű, Creative Commons Attribution 4.0 International License (CC BY 4.0) alatt jelent meg.
Magyar fordítás és szerkesztési megjegyzés: Magyar fordítás és ábraszöveg-adaptáció. A fordítás az eredeti szerzők, a forrás, a DOI és a licenc feltüntetésével készült; a változtatás a magyar nyelvű fordítás és szerkesztés.
Kapcsolódó tanulmányok a sorozatban: Botanikai-rendszertani modell 2020 · Genomikai-taxonómiai áttekintés 2023
Tartalomjegyzék
- Kivonat
- Főszöveg
- Módszerek
- Növényi anyag
- EH23 fázisolt, haplotípus-felbontású, kromoszómaléptékű horgonygenom
- EH23 F2 populáció
- EH23 F2 kannabinoid HPLC-módszerek
- EH23 RNS-szekvenálás
- EH23 haplotípus-expressziós elemzés
- Ace High ivar szerint torzított génexpressziós elemzés
- Hi-C könyvtárkészítés és szekvenálás
- HMW DNS izolálás és genomszekvenálás
- Pángenom-összeállítás és állványozás
- Referenciaalapú gráfépitás Minigraph-Cactus-szal
- Referenciafüggetlen gráfépitás PGGB-vel
- Vizualizáció és rövidolvasatok térképezése gráf-pángenomra
- Gráf-pángenom adatelérhetőség és metilált citozinok bázishívása
- Gén- és ismétlődés-előrejelzés
- Ideogrammódszerek
- Centromer- és telomeranalízis
- Riboszomális DNS detektálása és kvantifikálása
- Allélgyakorisági módszerek és PanKmer-genomelemzés
- Génalapú pángenom elemzése és gyűjtőgörbék
- K-mer elemzés és mag/dispensable gének azonosítása
- Ismétlődéselemzés
- SV-ket közvetlenül szegélyező TE-k és szinténiaelemzések
- Szelekció, TreeMix, lokális PCA és rezisztenciagén-analógok
- Terpén- és kannabinoid-bioszintézis gének azonosítása
- Szintáz-kazetta és kannabinoid-szintáz génanalízis
- K-mer kereszteződési elemzés és varin SNP asszociáció
- Ivari kromoszóma SDR-PAR határ azonosítása és összehasonlítása
- Jelentési összefoglaló
- Adatelérhetőség
- Kódelérhetőség
- Kiterjesztett adatok ábrái
- Köszönetnyilvánítás
- Tanulmány-információk
- Adat-, kód- és kiegészítő anyagok
- Magyar fordítás és szerkesztési megjegyzés
Kivonat
A Cannabis sativa globálisan fontos magolaj-, rost- és drogtermelő növényfaj. Egy évszázadnyi tilalom azonban súlyosan korlátozta a nemesítés és a csíraplazma-erőforrások fejlesztését, így a kenderalapú táplálkozási és rosthasznosítási lehetőségek nagyrészt kihasználatlanok maradtak. Itt egy kannabisz-pángenomot mutatunk be, amely 181 új és 12 korábban közzétett genomból épült, összesen 144 biológiai mintából, hím (XY) és nőivarú (XX) növényeket egyaránt beleértve.
A kannabisz-pángenom kiterjedt régióiban meglepően nagy, egyetlen fajhoz képest szokatlan diverzitást azonosítottunk, magas genetikai és strukturális változatossággal, és új populációs szerkezetet, valamint hibridizációs történetet javaslunk. Az ősi heteromorf X és Y ivari kromoszómákon változó határt figyeltünk meg az ivarmeghatározó és pszeudoautoszomális régiók között, valamint hímirányú expressziót mutató géneket, köztük több kulcsfontosságú virágzásszabályozót kódoló gént.
Ezzel szemben a kannabidiol-sav és a delta-9-tetrahidrokannabinol-sav előállításáért felelős kannabinoid-szintáz gének nagyon alacsony diverzitást tartalmaztak, annak ellenére, hogy egy változékony régióba ágyazódnak, több pszeudogenizált paralóggal, strukturális variációval és eltérő transzponálhatóelem-elrendezésekkel.
Emellett olyan acil-lipid-tioészteráz génváltozatokat azonosítottunk, amelyek összefüggtek a zsírsavlánc-hossz változatosságával, valamint a ritka kannabinoidok, a tetrahidrokannabivarin és a kannabidivarin termelésével. Következtetésünk szerint a C. sativa génkészlete továbbra is csak részben ismert, az ázsiai vad rokonok létezése valószínű, és a faj kultúrnövényi potenciálja nagyrészt kihasználatlan.
Főszöveg
Bevezetés
A kannabisz (C. sativa L., kannabisz) ősi domesztikált növény, amelynek mag- (kaszat-) és rosthasznosítására Kelet-Ázsiában 8000 évvel ezelőttről széles körű régészeti bizonyíték áll rendelkezésre, korábbi előfordulásai pedig akár 12 000 évre nyúlnak vissza [1,2]; ez a fontos kultúrnövények, például a búza, árpa, kukorica és rizs koraiságával vetekszik. A kannabisz eredetileg többhasznú kultúrnövény volt Ázsiában, ahol ugyanazokat a növényeket rost-, élelmiszer- és drogforrásként hasznosították [2,3].
Az idők során a kannabisz globálisan elterjedt, és egy- vagy kettős hasznosítású kultivárok alakultak ki, végül létrehozva a huszadik század eltérő kender- és drogtípusú populációit [4]. Az 1900-as évek eleje előtt a kannabisz fontos árucikk volt Ázsiában, Európában és az Újvilágban, és vitorlákhoz, kötelekhez, ruházathoz és papírhoz használt rostokat állítottak elő belőle. Más rostnövények versenye, a drogtörvényekkel való összefonódás, majd a szintetikus rostok kifejlesztése azonban a termelés visszaeséséhez vezetett. Az utóbbi évtizedekben a kannabisz használata specializált alkalmazások felé tolódott el, ideértve a réspiacot jelentő magolajokat és a drogtermelést, ahol napjainkban is jelentős gazdasági és kulturális fontossággal bír [5].
A történelem során és világszerte a kannabisz a „termesztés, fogyasztás és fellépés” ciklusain ment keresztül [6]. A modern tilalom az Egyesült Államokban, a huszadik század elején alakult ki [7], de 1961-re a legtöbb országra kiterjedt [8]. A tilalom évtizedekre megszüntette a kannabisz rost- és élelmiszerhasználatát, ugyanakkor nagy értékű illegális piacot hozott létre a mirigyszőrökből származó fitokannabinoid-alapú drogok számára.
Bár több mint 100 fitokannabinoidot azonosítottak, csak korlátozott számú termelődik jelentős mennyiségben; ezek alapján sorolják a növényeket kemotípusokba: delta-9-tetrahidrokannabinol-sav (THCA; I. típus), kannabidiolsav (CBDA; III. típus), kiegyensúlyozott CBDA és THCA (II. típus), kannabigerolsav (CBGA; IV. típus), valamint kannabinoidmentes (V. típus) [9]. Noha a tetrahidrokannabinol (THC), az elsődleges bódító hatású vegyület, továbbra is ellenőrzött anyag, az Egyesült Államok tagállamainak többsége és számos ország már engedélyezi a kannabisztermékek orvosi vagy felnőttkori használatát. Külön folyamatként az USA 2014-es és 2018-as Farm Bill törvényei elősegítették a 0,3% alatti THC-t termelő növények kendertermesztését és kutatását az USA területén, lehetőségeket teremtve a nem THC-alapú drog-, gabona- és rostalkalmazások fejlesztésére.
A haploid kannabiszgenom viszonylag kis méretű (körülbelül 750 Mb), összetettségét azonban a transzponálható elemek (TE-k) magas aránya (körülbelül 79%) és a jelentős heterozigozitás (egynukleotidos polimorfizmusok, SNP-k: több mint 2%) adja.
A jól ismert, epilepsziaellenes „Charlotte’s Web” kultivárral rokon, magas kannabinoid-tartalmú (HC) kannabidiol- (CBD-) kender leszármazási vonalból származó CBDRx (cs10) referenciagenom a kannabinoid-szintáz gének elrendezését egyetlen teljes hosszúságú CBDAS-kópiaként oldotta fel, amely konzervált, 70-80 kb méretű tandem TE-tömbökbe ágyazódik [10]. Továbbá a CBDRx-hez hasonló HC kender vonalak a CBDAS lókusz döntően marihuána (MJ) genetikai háttérbe történő introgressziója révén jöttek létre, ezáltal nagy hatáserősségű allélokat hasznosítva a CBD-termelés fokozására [11]. A közzétett kannabiszgenomok kezdeti összehasonlítása ugyanakkor a hasznosítási típusok között jelentős genomi dinamizmust jelez [11-16], ami kulcsfontosságú, továbbra is megválaszolatlan kérdéseket vet fel a genetikai diverzitás globális kiterjedéséről.
Ezenfelül továbbra sem világos, milyen szerepet játszott a hibridizáció a genomarchitektúra és az allélátadás alakításában, ami további kiváló minőségű összeállítások és populációléptékű genomikai elemzések szükségességére mutat rá. Itt átfogó keretet hoztunk létre e többhasznú kultúrnövény genetikai diverzitásának feltárására azáltal, hogy haplotípus-felbontású, kromoszómaléptékű összeállításokkal kannabisz-pángenomot készítettünk.
A kannabisz-pángenom
A kannabiszt gyakran monospecifikus nemzetségként osztályozzák [17], bár továbbra is vita tárgya a Cannabis indica Lam. és a Cannabis ruderalis státusza; utóbbit a nappalhossz-semleges (DN; autoflowering) virágzási típus forrásának tartják [18]. A kannabisz diverzitását úgy közelítettük meg, hogy a pángenomhoz több forrásból választottunk mintákat, lefedve a hasznosítási típusokat, történeti hátteret, ivarkifejeződést és agronómiai tulajdonságokat (Kiterjesztett adatok 1. ábra és 1. kiegészítő ábra).
A kannabisz-pángenom 181 új PacBio-összeállításból és 12 korábban publikált genomból áll, 144 biológiai mintát képviselve; ezek között 78 haplotípus-felbontású, kromoszómaléptékű összeállítás és 103 kontigszintű összeállítás található. Kiemelünk egy F1 hibridet (ERBxHO40_23; EH23), amely két fenotípusosan és genetikailag eltérő szülő között jött létre; ez segít tisztázni a genom olyan jellemzőit, amelyek korábbi vizsgálatokból hiányoztak (1a. ábra, Kiterjesztett adatok 2. és 3. ábra, valamint 1. kiegészítő jegyzet).
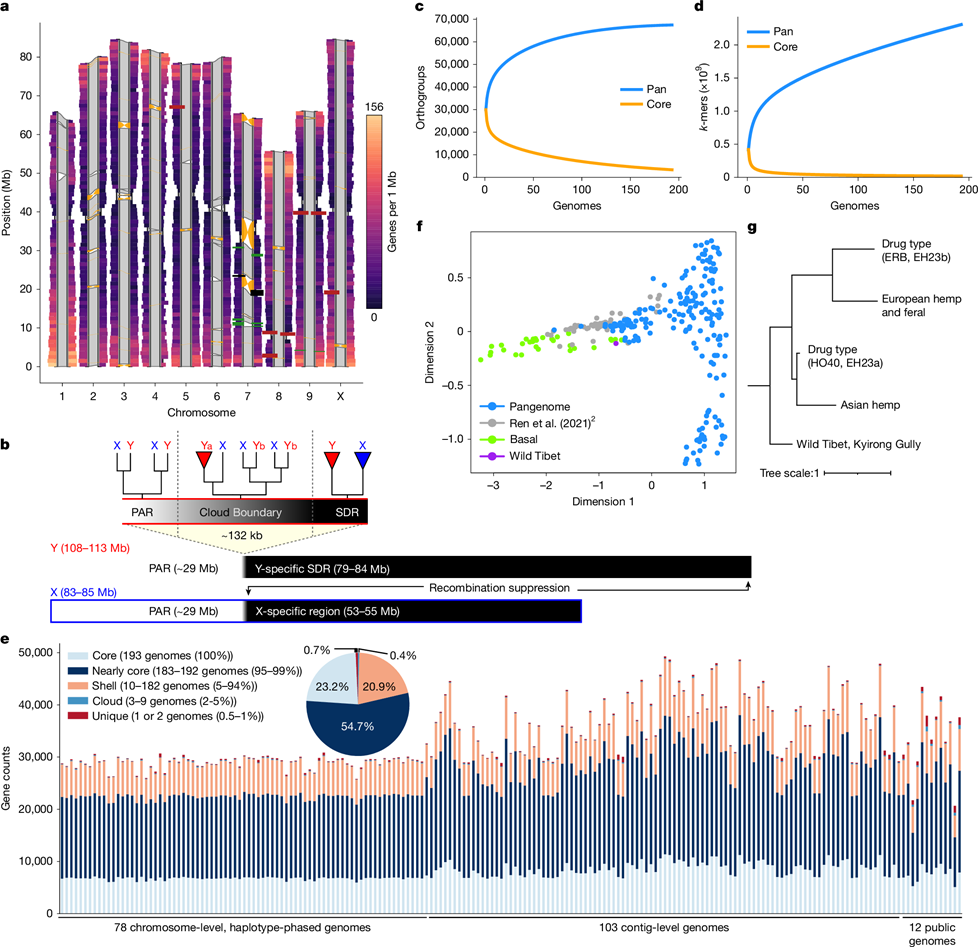
Minden genom jó minőségű volt: az átlagos N50 érték 7,5 Mb, a BUSCO [19] szerinti genom- és proteomteljesség pedig 97%, illetve 95%. Az átlagos haploid genom-hossz 781 Mb volt, genomonként körülbelül 35 000 fehérjekódoló génnel. A döntően idegentermékenyülő viselkedéssel összhangban az SNP-alapú heterozigozitás 1% és 2,5% között változott.
Az összeállítások szerkezetileg is kiváló minőségűek: megoldották a korábbi TE-elhelyezési problémákat, és feltárták a centromerrégiókat, telomerhosszt, nagy strukturális variációkat (SV-k), fontos gének – például a kannabinoid-szintázok – finomléptékű genetikai architektúráját, valamint az Y kromoszóma ivarmeghatározó régióját (SDR) és pszeudoautoszomális régióját (PAR), amely a kannabiszgenom legnagyobb kromoszómája.
Referenciaalapú és referenciafüggetlen megközelítésekkel egyaránt átfogó Cannabis-pángenomokat hoztunk létre. Referenciaalapú pángenomgráfot a Minigraph-Cactus (MGC) [20] segítségével készítettünk a 78 kromoszómaléptékű, haplotípus-felbontású genomból. Referenciafüggetlen megközelítésként a PanKmer [21] segítségével mind a 193 genomból k-mer mátrixot, a PanGenome Graph Builder (PGGB) [22] segítségével pedig gráfalapú reprezentációt építettünk.
A PGGB nagy memóriaigénye miatt a gráfgeneráláshoz 16 genom részhalmazát választottuk. Az MGC és a PGGB által detektált SV-k szorosan megfeleltek a páronkénti teljesgenom-illesztésekből származó eredményeknek. Egy változatos rövidolvasat-adatkészlet leképezési aránya hasonló volt az MGC pángenomgráf (95,09%) és a lineáris EH23a referenciagenom (95,0%) esetében, ami azt jelzi, hogy mindkét megközelítés hatékonyan ragadta meg a variációt.
A pángenom öt populációt tár fel
A kannabisznemzetség taxonómiájáról, történetéről és nevezéktanáról régóta vita folyik [23]. Széles fenotípusos és földrajzi diverzitása miatt vagy több fajból álló, egymással kereszteződő komplexként, vagy egyetlen fajként, alfaji besorolásokkal osztályozták. A pángenom teljességének és diverzitásának értékeléséhez a gyűjtőgörbét megosztott génalapú ortocsoportok és megosztott k-merek alapján számítottuk ki (1c,d. ábra).
A görbe azt jelezte, hogy a kannabisz ortocsoport-diverzitásának többségét körülbelül 100-125 genomnál megragadtuk, jóllehet jelentős globális genomi változatosság maradt jellemezetlenül, feltehetően a közelmúltbeli TE-aktivitás miatt. A 78 haplotípus-felbontású, kromoszómaléptékű összeállítás gyűjtőgörbéi hasonló, de mérsékeltebb diverzitás-mintaszám összefüggést mutattak.
Az összes pángenom-mintában azt találtuk, hogy a gének 23%-a „mag” gén (minden genomban jelen van), 55%-a „majdnem mag” gén (a genomok 95-99%-ában jelen van), 21%-a „héj” gén (a genomok 5-94%-ában van jelen), míg egy kis hányad „felhő” (0,4%) vagy „egyedi” (0,7%) kategóriába tartozik. A terpén-bioszintézissel és védekezési válasszal kapcsolatos génontológiai (GO) kifejezések a maggének között leggyakrabban gazdagodó kategóriák közé tartoztak, bár mindkettő jelentős szekvenciaszintű variációt mutatott.
A kannabiszban az ősi lambda esemény óta, körülbelül 100 millió éve nem történt teljesgenom-duplikáció [13]. Ez arra utal, hogy kiterjedt genomi diverzitása nem közelmúltbeli teljesgenom-duplikációkból vagy hibridizáció által hajtott allopoliploidiából, hanem tandem gén-duplikációból és más lokális duplikációs mechanizmusokból ered.
A populációk közötti összehasonlítások, amelyeket fázisolt SNP-k alapján páronkénti átlagos Fst (fixációs index) értékekkel végeztünk, azt mutatták, hogy egyes kannabiszpopulációk genetikai differenciálódása fajok közötti összehasonlításokhoz, például a szamócában megfigyelt értékekhez hasonló szintet ért el [24] (Fst = 0,20 az MJ és a kender között).
A magas Fst-értékű SNP-ket hordozó konkrét gének környezeti válaszokhoz kapcsolódtak; a cirkadián, fényjelátviteli és virágzási idő gének átlag feletti Fst-értéket (0,42) mutattak. Különösen figyelemre méltó, hogy a GIGANTEA (GI) [25], egy erősen konzervált, rendszerint egykópiás gén, amely központi szerepet játszik a napi periódushosszt, virágzási időt és sejtmegnyúlást szabályozó cirkadián órában, az ötödik legmagasabb Fst-értékű SNP-t tartalmazta (0,77, MJ kontra kender).
Külön elemzésben, 20 kb-os SNP-ablakokon végzett szelektív söprés teszttel (XP-CLR, MJ kontra kender) a GI ismét az X kromoszóma egy szignifikáns régiójába esett. A géncsalád-diverzitás tágabb elemzése jelentős variációt tárt fel a GI lókusznál a HC kender és a kender populációk között. Ezek az eredmények kiemelik a szelekció hatását olyan kulcsfontosságú agronómiai génekre [26], amelyek a virágzás és az internódium-megnyúlás (rosthossz) eltérései mögött állhatnak, és amelyek erősen különböznek a kender- és MJ-populációk között.
Az Észak-Amerikában magas kannabinoidszintet termelő drogtípusú populációkról úgy tartják, hogy Délkelet- és Közép-Ázsia régióiból erednek, és a Karib-térségen, valamint Dél-Amerikán keresztül jutottak a nyugati féltekére; e feltételezett ősi populációkról azonban a rendelkezésre álló ismeretek többsége korlátozott történeti beszámolókon és feltételezéseken alapul [5].
A PanKmer-pángenom k-mer alapú hierarchikus klaszterezése a drogtípusú minták két tág csoportját javasolta: az egyik az ázsiai kenderhez, a másik az európai kenderhez igazodott (1f,g. ábra). Mindkét csoport tartalmazott MJ és HC kender mintákat; utóbbiakról úgy gondolják, hogy döntően MJ-származásúak, közelmúltbeli, CBDAS génekre irányuló introgressziós nemesítési múlttal, talán európai kender eredetből [11].
Amikor azonban fázisolt SNP-alapú szerkezetet alkalmaztunk úgy, hogy az összes MJ mintát egy populációként kezeltük, a TreeMix modell legnagyobb valószínűségű filogenetikai fát becsült, hat génáramlási (migrációs) eseménnyel az ázsiai kender, HC kender és európai kender, valamint az MJ és HC kender minták között. Ezek az eredmények részben magyarázhatják a drogtípusú minták európai és ázsiai csoportosulását a k-mer klaszterezésben, és tükrözhetik az ázsiai és európai kender történetileg dokumentált hibridizációs nemesítésének hatását [27].
A két drogtípusú populáción, valamint a külön európai és ázsiai kender populációkon túl a k-mer klaszterezés jelentős divergenciát mutatott az egyetlen rendelkezésre álló vad tibeti összeállítás és minden más domesztikált vagy elvadult vonal között [13], ami arra utal, hogy Ázsia távoli régióiban még létezhetnek vad Cannabis rokonok [2].
Valóban, a pángenom-összeállítások és az Európából, illetve Ázsiából gyűjtött minták rövidolvasatainak együttes k-mer alapú hierarchikus klaszterezése visszaadta az eredeti szerzők megállapítását, miszerint az Ázsiából származó, „drogtípusú elvadult” és „bazális” minták elkülönülő populációkat képviselnek [2]. Végső soron a domesztikációra, biogeográfiára és hasznosítási típus történetre vonatkozó hipotézisek finomításához az ázsiai és történeti példányok szélesebb mintavétele, valamint a vad és elvadult populációk gondos elhatárolása szükséges.
Az ivari kromoszómák evolúciója
A kannabisz ivarkifejeződése régóta foglalkoztatja a biológusokat [28]. Bár a legtöbb populáció kétlaki, külön hím (XY) és nőivarú (XX) növényekkel, egylaki (XX) formák is léteznek, amelyek változó arányban hoznak hím és női virágokat. A Cannabaceae ivari kromoszómái a Cannabis és a Humulus közös ősében, több mint 36 millió éve (Ma) keletkeztek [29] – korábban, mint a korábbi becslések [30] -, így a virágos növények legrégebbi ismert ivari kromoszómái közé tartoznak [31].
Ősi eredetük ellenére a kannabisz ivari kromoszómáit az ivari dimorf tulajdonságokra irányuló emberi szelekció is alakította [32]. Drogtípusú populációkban a hímek kevés mirigyszőrt termelnek, a megporzás pedig csökkenti a nőivarú növények kannabinoidhozamát, ami a hímek nemesítési programokban való használatának csökkenéséhez vagy megszüntetéséhez vezetett. Ezzel szemben a kendermag-termeléshez pollen szükséges, a hím növények pedig javítják a háncsrosthozamot és -minőséget. Emellett az európai egylaki rostkultivárokat, például a Santhicát (SAN) és a KC Dorát (KCDv1), a rost és mag gépi betakarítási hatékonyságának javítására fejlesztették ki, újabb mesterséges szelekciós réteget adva hozzá [31].
A legtöbb zárvatermővel ellentétben a kannabisznak heteromorf XY párja van; az Y kromoszóma körülbelül 30%-kal nagyobb, mint az X kromoszóma. A rekombináció a PAR-ban zajlik, az Y kromoszóma SDR régiójában azonban gátolt. Az SDR a körülbelül 110 Mb méretű Y kromoszómából 79-84 Mb-ot fed le, így a növények egyik legnagyobb SDR-je, 840-1160 génnel. Ezzel szemben a PAR csak körülbelül 29 Mb, mégis 1900-1980 gént hordoz, köztük számos fontos virágzási gént, például a FLOWERING LOCUS T (FT), CONSTANS (CO) és GI géneket.
Az elmélet azt jósolja, hogy a rekombináció kezdeti gátlása után az SDR lépcsőzetesen tágul, mivel a szelekció a hímek számára előnyös, de a nőstények számára hátrányos géneket kapcsol az SDR-hez [33]. Alternatívaként semleges folyamatok is vezérelhetik az SDR tágulását, amit a szinonim szubsztitúciós ráták (Ks) tükröznek. Az SDR menti Ks-értékek folyamatos génhozzáadási mintázatot mutattak a PAR-határtól a centromer felé [29], ami arra utal, hogy a centromer közelében fellépő rekombinációgátlás legalább részben hozzájárult a táguláshoz.
K-merek és X-Y ortológ filogéniák segítségével két eltérő SDR-haplotípust azonosítottunk: a Ya-t, amelyet hat minta osztott meg, és a Yb-t, amely két mintában fordult elő (1b. ábra). E haplotípusok az SDR-PAR határnál különböztek: öt konzervált génmodell választotta el őket, nagyjából 51 kb-tól (GVA-21-1003-002) [34] 132 kb-ig (Kompolti), míg minden más eset 61-62 kb-t fogott át.
A Ya haplotípusban a PAR-SDR határhoz legközelebb elhelyezkedő gén (amely a Yb-ben a PAR-ban található) a TRANSCRIPTION ELONGATION FACTOR (SPT5), amelyről ismert, hogy Arabidopsisban a hidegindukált virágzás során a FRIGIDA révén kölcsönhatásba lép a FLOWERING LOCUS C (FLC) génnel [35]. Ez arra utal, hogy a virágzási idő génekre irányuló szelekció elősegítette a rekombinációgátlás lépcsőzetes eltolódását és az SDR tágulását; ez magyarázhatja, hogy egyes fajtákban a hímvirág-fejlődés a nőivarú virágzás kezdete előtt indul.
Az SDR-PAR határ polimorfizmusai jelzik, hogy a kender génkészletében fennmaradt a szexuálisan antagonisztikus gének ősi diverzitása, amely a virágzási időzítés hasznos változatossága mögött állhat [36]. Ezenkívül az Ace High (AH3M) hím és női szöveteinek génexpressziós profilozása több mint 7000 gén hímvirágban torzított expresszióját tárta fel az összes kromoszómán, számos funkciót lefedve, köztük a pollenfejlődést. Ez ellentétben állt a hím levélben (körülbelül 1400 gén), női levélben (körülbelül 3700 gén) és női virágban (körülbelül 3900 gén) torzított expressziót mutató génekkel.
Míg az X kromoszómán a génexpresszió meglehetősen egyenletes volt, az Y kromoszómán a génsűrűség és expresszió a PAR felé tolódott. Figyelemre méltó, hogy a PAR génjeinek jelentős hányada (38%, körülbelül 750 gén) hímvirág-torzított expressziót mutatott, szemben az SDR 6%-ával (94 gén). Bár az SDR egy vagy több, hímvirág-fejlődést meghatározó, eddig azonosítatlan ivarmeghatározó gént kódol, a hím vagy női virággén-expresszióhoz szükséges transzkripciós hálózat döntő része szélesen, valamennyi kromoszómán eloszlik.
A transzponálható elemek alakítják a pángenomot
A transzponálható elemek (TE-k) meghatározó szerepet játszottak a kannabiszgenom alakításában, különösen az intronmentes kannabinoid-szintáz gének felszaporodásában, amelyek 70-80 kb méretű konzervált TE-kazettákba ágyazódnak [11]. Átlagosan a TE-k minden genom 68%-át tették ki, a hosszú terminális ismétlődésű retrotranszpozonok (LTR-RT-k) pedig az összes 50%-át képviselték (2a. ábra). A gének átlagosan TE-k közelében helyezkedtek el (443-613 bp távolságra).
A különböző TE-típusok eltérő beékelődési mintázatot mutattak: a DNS-transzpozonok (például Mutator és Helitron) a kódoló régiók előtt 500 bp-on belül ékelődtek be, míg az LTR-RT-k egyenletesebben oszlottak el a gének két oldalán. A transzpozícióban, transzkripcióban, rekombinációban és DNS-javításban részt vevő gének gyakran Ty3-LTR-ekkel társultak, míg a védekezési és metabolit-bioszintézis gének Ty1-LTR-ek közelében gazdagodtak.
Számos intakt TE-ről becsültük úgy, hogy az elmúlt 100 000 évben épült be a genomba, ami azt sugallja, hogy a folyamatban lévő diverzifikációt hibridizáció és stressztényezők hajthatják, különösen az F1 és MJ populációkban. Az egyik ilyen tényező a klonális szaporítás, amely a modern MJ-termelésben gyakori, de a szántóföldi kendertermesztésben ritka.
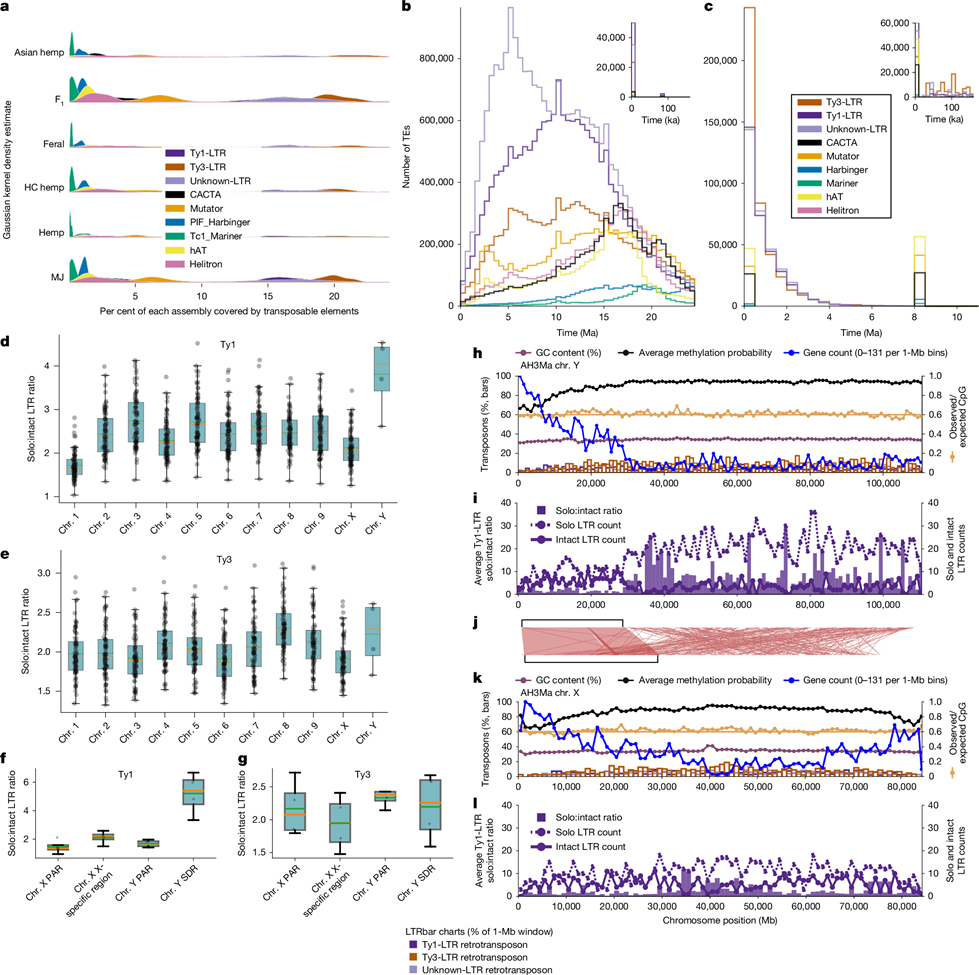
A 4 millió éven át fennmaradó aktivitás és az LTR-felszaporodás közelmúltbeli fellángolása ellenére a kannabiszgenom megőrizte kisebb haploid genom-méretét (körülbelül 750 Mb) testvérnemzetségéhez, a Humulushoz képest, amely a Humulus japonicus 1700 Mb-jától a Humulus lupulus 2700 Mb-jáig terjed [37]. A solo LTR-ek a genom tisztulását tükrözik, és ektopikus rekombinációval alakulhatnak ki, amely a teljes LTR-RT belső szekvenciájában történik [38].
A kannabiszban megfigyelt magas solo:intakt arány valószínűleg hozzájárul kompakt genom-méretéhez a TE-felhalmozódás mérséklésével. A Ty1-LTR-ek a legmagasabb solo:intakt arányt az Y kromoszóma SDR régiójában mutatták, ami arra utal, hogy e régió kezdeti tágulását TE-beékelődések hajtották, amelyeket ektopikus rekombinációval történő törlési események előztek meg vagy követtek. A DNS-metiláció szintén gátolja a kontrollálatlan TE-felszaporodást az expresszió elnémításával [39].
A TE-metilációs szintek magasabbak voltak a genomszintű átlagoknál, bár populációspecifikus különbségeket észleltünk. Az EH23 F1 hibridben expresszált TE-transzkriptumokat mutattunk ki, ami folyamatos TE-aktivitást jelez. Az Y kromoszómán a PAR és az SDR eltérő génexpressziós és intakt TE-expressziós mintázatot mutatott, az SDR fokozott metilációs szintjeivel, összhangban degenerált, génszegény természetével.
Több TE-család aktívan transzkribálódik, és sok beékelődés evolúciósan közelmúltbeli; a TE-gyakorisági profilok ugyanakkor populációnként jól elkülönülő mintázatot mutattak. Bizonyos TE-típusok közelmúltbeli divergenciaideje, génekhez közeli gazdagodása és populációspecifikus eloszlása együtt arra utal, hogy a TE-k hozzájárulnak mind a génevolúcióhoz, mind az adaptív válaszok szabályozásához a kannabiszban.
A strukturális variánsok innovációt hajtanak
A kannabiszban fiatal, aktív TE-k nagy bősége miatt megvizsgáltuk szerepüket a pángenom strukturális variánsainak (SV-k) alakításában (3. ábra). Az SV-számok leginkább a transzlokációkban és duplikációkban változtak, tükrözve a populációspecifikus TE-bőséget, míg az inverziók mutatták a legkisebb variációt (átlagosan 86 genomonként).
Az inverzióméretek azonban 200 bp-tól 25 Mb-ig terjedtek (átlag 304 kb), multimodális eloszlást alkotva; ez arra utal, hogy különböző hosszúságú inverziókat többféle evolúciós erő alakított. Míg az SNP-heterozigozitás a pángenomban 1 és 2,5% között mozgott, az SV-ket és nem illeszthető régiókat is beszámító heterozigozitás (változó régiók) átlagosan a teljes genom-hossz 20,6%-a volt, ami rávilágít a kannabisz korábban jellemezetlen genomi változatosságának mértékére.
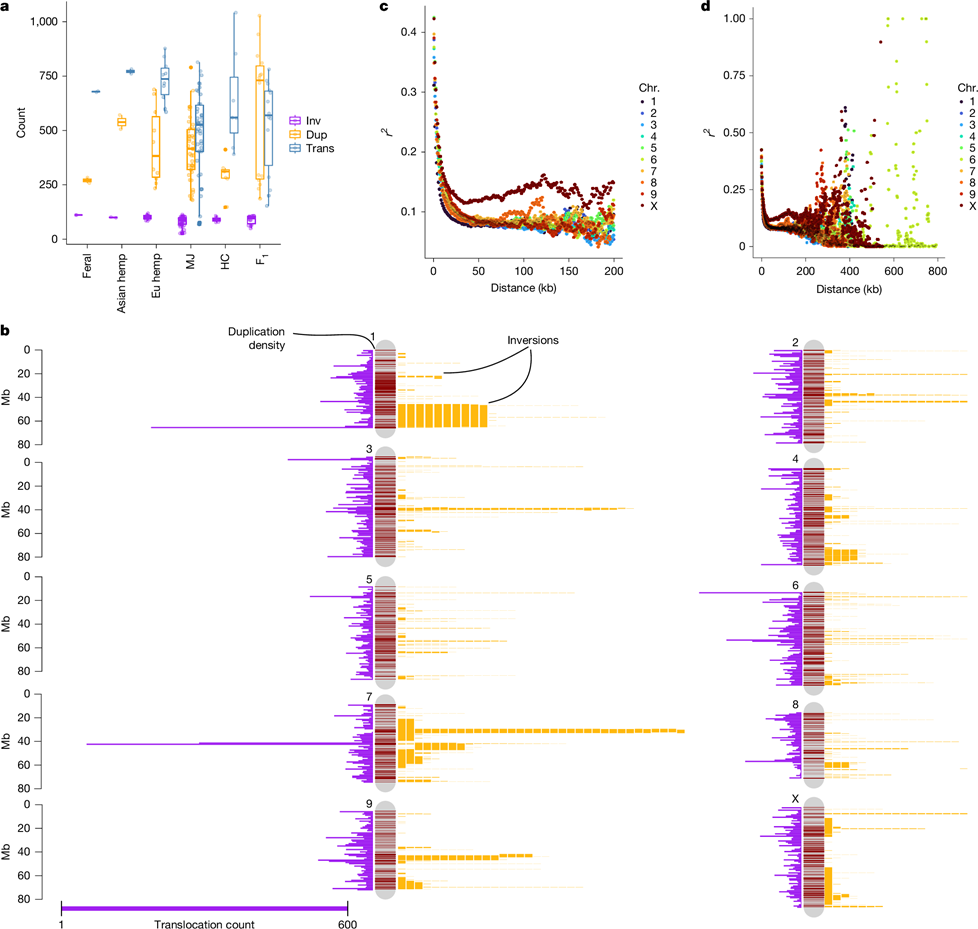
A TE-k gyakran okoztak kis- és közepes méretű transzlokációkat, duplikációkat és inverziókat, míg a nagyobb inverziók olyan töréspontokon jöttek létre, amelyek szegmentális duplikációkban és fordított ismétlődésekben gazdagodtak [40]. Az 1., 4. és 7. kromoszómán lévő SV-forrópontok közös inverziós töréspontokkal és TE-gazdag régiókkal fedtek át. Az SV-töréspontokban (500 bp felfelé és lefelé, összesen 1 kb) található TE-k elemzése populációspecifikus TE-gazdagodási mintázatokat tárt fel.
Az MJ-genomokban a duplikációk gyakran három DNS-TE családot és Ty3-LTR-RT-ket tartalmaztak (P < 0,05, Welch-féle t-próba). Más populációkban csak a Harbinger és Mutator DNS-TE-k gazdagodtak a duplikációs töréspontoknál, míg az elvadult kender duplikációi nem mutattak szignifikáns TE-gazdagodást; ez közelmúltbeli TE-aktivitásra vagy alternatív SV-képződési mechanizmusokra utal.
Az inverziók a genom akár 7%-át is lefedték, meghaladva a többfajos összehasonlításokban – például a szójában és szőlőben – megfigyelt értékeket [41]. A TE-k és SV-k populációspecifikus kölcsönhatása, valamint gyakori génközelségük alapján eredményeink a kannabiszgenom evolúcióját hajtó mechanizmusok változatos készletét tárták fel, amelyek közül sok a korábbi újraszekvenálási erőfeszítésekben rejtve maradt.
A kannabiszgenom több régiójában megfigyeltek szegregációs torzulást [16], ami tükrözi az F1 EH23 hibridben észlelt mintázatokat, és arra utal, hogy az SV-k hozzájárulhatnak az allélátadási torzulásokhoz [42]. A hosszú inverziók, például az 1. kromoszómán található 19,5 Mb hosszú inverzió, szupergénként működhetnek, amelyet esetleg asszociatív túldominancia révén fenntartott kiegyensúlyozott polimorfizmus őriz meg [43].
Ezen inverzió 17 előfordulása közül 15 mintában heterozigóta, egyben homozigóta állapotot találtunk. Az invertált régió körülbelül 1203 gént tartalmazott, sokféle funkcióval, köztük a cirkadián és virágzási idő maggénjével, a PSEUDO RESPONSE REGULATOR 3-mal (PRR3). A PRR3 kapcsolatba hozható a kannabisz „autoflower” DN viselkedésével [44], valamint a nagy kultúrnövények (szója és cirok) területkiterjedéséhez és természetes populációkhoz kapcsolódó virágzási idő-variációval [45-47].
A PRR3 egy magas Fst-értékű SNP-t (0,61), továbbá torzított expressziót tartalmazott az F1 EH23 hibridünkben, amely recesszív volt a DN tulajdonságra. A páronkénti SNP r2 értékek és a lokális főkomponens-elemzés (PCA) ábrái e régióban bizonyos fokú haplotípusképződést és fokozott kapcsoltsági egyensúlyhiányt (LD; >10 kb) jeleztek, különösen a belső töréspontnál. Ezek azonban nem mutatták a teljes differenciálódás vagy rekombinációgátlás egyértelmű jeleit, ahogyan azt más fajokban kimutatták [48].
Domesztikált kannabinoid-útvonal
A kannabisz az egyetlen bőséges kannabinoid-termelő növény, bár más növények (például májmohák) és gombák kisebb mennyiségeket szintetizálnak [49]. Bár a kannabinoid-bioszintetikus útvonal kulcsenzimjeit már azonosították (4a. ábra), az útvonal utolsó lépésének genomi szerveződése a kannabiszgenom komplexitása miatt megoldatlan maradt.
E rejtély tisztázódott, amikor teljes hosszúságú THCAS, CBDAS és CBCAS géneket fedeztek fel konzervált TE-kazettákba ágyazva, a 7. kromoszómán tömbökbe rendeződve [11]. Nem volt azonban világos, hogy a szintáz géneknek ez a TE-közvetített elrendezése mennyire konzervált a kannabisz-pángenomban.
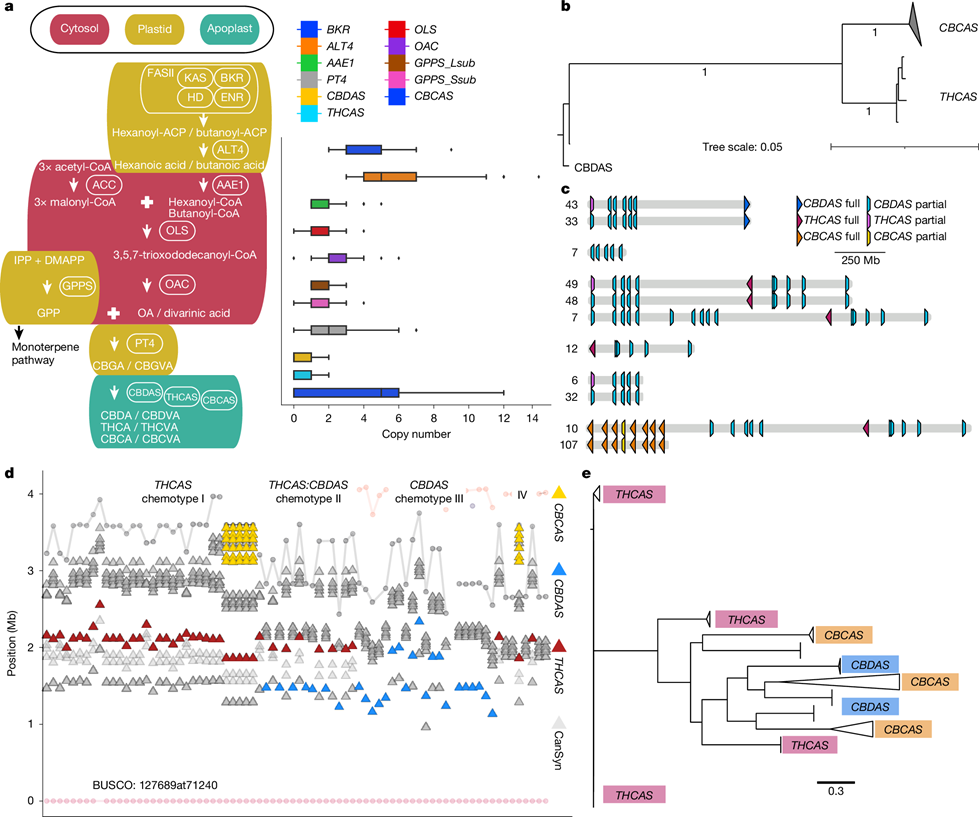
A kannabinoid-szintázok az ősi Berberine bridge enzyme-like (BBE-like) géncsaládból duplikálódtak és neofunkcionalizálódtak a 7. kromoszómán, majd a domesztikációs folyamat során végül a funkcionális THCAS és CBDAS allélok korlátozott készletére redukálódtak [11,50].
A pángenomban minden haploid genom legfeljebb egy teljes hosszúságú THCAS vagy CBDAS gént hordozott, amelyek hasonló TE-kazetta-tömbökbe rendeződtek; ezek többsége szintáz pszeudogéneket tartalmazott. E kannabinoid-szintáz-kazetták korlátozott számú elrendezésben fordultak elő, specifikus TE-khez kapcsolódva, ami arra utalt, hogy a szelekció a funkcionális allélok szűk körét pszeudogén kazetta-haplotípusokhoz kapcsolta.
Ennek eredményeként a legtöbb THCAS és CBDAS gén nem szinténikus volt, és a kannabisztípusok közötti inverziókkal társult, de általában a 7. kromoszómán nagyjából 1,5 Mb-ra korlátozódó régióban helyezkedett el. Miközben a kannabisz-pángenom nagy genomi variációt mutat, a THCAS és CBDAS lókuszok konzervált szerkezete arra utal, hogy e régiók erős szelekciós nyomás alatt állnak.
A teljes hosszúságú CBCAS paralógok rendszerint 15-20 Mb-ra voltak a 7. kromoszóma centromerétől, de genomi inverzió miatt néha körülbelül 1,2 Mb-ra kerültek a THCAS-tól. A CBCAS a genomok 56%-ában (110/193) fordult elő, 1-15 kópiás tömbökben. Bár a CBCAS képes kannabikroménsavat (CBCA) termelni élesztőben [16], több mint 59 000 kannabiszminta elemzése szinte semmilyen CBCA-t nem észlelt, valószínűleg a természetes alacsony szintek miatt [51].
Az EH23-ban a CBCAS-expresszió minden szövetben alacsony volt, ami azt sugallja, hogy a CBCA-felhalmozódás nem állt erős szelekció alatt, potenciálisan az emberi THC- és CBD-preferencia miatt.
Varin kannabinoidok és zsírsavgének
A növényben termelődő kannabinoidok alkil oldalláncának hossza egytől legalább hét szénatomig változhat, a modern génkészletekben az öt szénatom a leggyakoribb [52]. A három szénatomos oldalláncú kannabinoidok (propil; tetrahidrokannabivarin, THCV; kannabivarin, CBDV; kannabigerovarin, CBGV) jóval ritkábbak, de új terápiás ágensekként érdeklődést váltottak ki [53].
Korábbi vizsgálatok jellemezték e tulajdonság poligénes természetét, és a β-keto-acil-hordozófehérje-reduktáz (BKR) gént a varin kannabinoidok termelésével hozták összefüggésbe, de legalább egy lépést nyitva hagytak a teljes bioszintetikus hipotézishez [54]. A varin kannabinoid-termelés modelljét azáltal bővítettük, hogy azonosítottuk az acil-lipid-tioészteráz (ALT3 és ALT4) gének komplexét a 7. kromoszóma eleje közelében, amely az F2 térképezési populációnkban a varintermeléssel társult, és k-mer alapú trio-keresztezési elemzésünkben közös haplotípuson belül helyezkedett el.
A kannabiszban magas ALT génkópiaszám-változatosságot találtunk, amely mind fázisolt, mind fázisolatlan összeállításokat figyelembe véve 2-14 kópia között mozgott, 4 kromoszómán. A legtöbb növényi genomban 4-5 ALT homológ található, egyesekben pedig csak egyetlen homológ (például Brassica rapa és Glycine max) [55]. A kannabisz ALT fehérjeszekvencia-változatossága szintén figyelemre méltó volt: az EH23a és EH23b genomokban minden ALT4 eltérő ortocsoport-tagságot mutatott, noha e gének hasonló pozíciókban helyezkedtek el.
Mivel a növényi zsírsav-acil-tioészterázok legrövidebb ismert zsírsavterméke egy 6:0 zsírsav, amelyet az Arabidopsis ALT4 állít elő, az EH23a ALT4 allél vezető jelölt további kísérletekre. A kereszteződési helyek, a kapcsoltsági egyensúlyhiány lehetősége és e régió rövidolvasat-leképezési problémái miatt azonban bármelyik ALT3 vagy ALT4 transzduplikált gén (vagy splicing variáns) lehet oksági a varin kannabinoid-termelésben. Alternatívaként részben átfedő, szubfunkcionalizált szubsztrátspecificitásuk lehet, ami kihívást jelent a további térképezési és fejlesztési erőfeszítések számára [56].
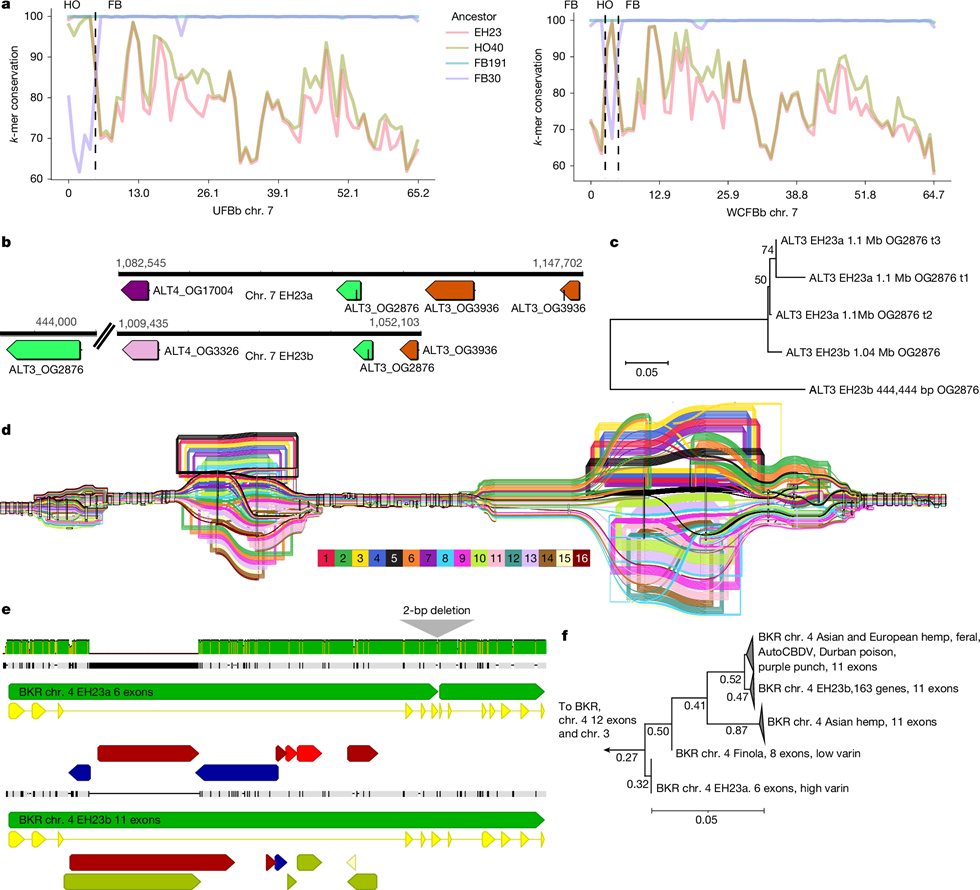
Bár a 4. kromoszómán lévő BKR gént korábban egy teljesgenom-asszociációs vizsgálat azonosította, a pángenom kimutatta, hogy egy 2 bp-os deléció 6 exonos funkcióvesztett génmodellt hozott létre, amelyből hiányoztak a katalitikus aktívhely-maradékok. Így e gén funkciójának csökkenése vagy elvesztése valószínűleg szükséges a butiril-acil-hordozófehérje-készlet növeléséhez, amelyet ezt követően valamelyik ALT3 vagy ALT4 géntermék hidrolizál vajsavvá, elvezetve a varin kannabinoid bioszintéziséhez.
Mivel a kannabisz a 3. és 4. kromoszómán is hordoz BKR géneket, az egyik kópia katalitikus funkciójának elvesztése valószínűleg nem szünteti meg teljesen az iteratív zsírsavlánc-szintézist; ez azt is magyarázhatja, miért találhatók a varin kannabinoidok csak bizonyos arányokban a pentil kannabinoidokkal [52,54].
A pángenomban az EH23a 6 exonos BKR variáns kizárólag a HO40 pedigré mintákban fordult elő (magas varin); minden más minta, egy alacsony varintermelő Finola magolaj-kultivár 8 exonos BKR-verzióját kivéve, 11 vagy 12 exonos modelleket hordozott. Az előre jelzett BKR fehérjék filogenetikai viszonyai szerint a 6 exonos gén közelebb állhat bizonyos ázsiai kender, európai kender és elvadult variánsokhoz.
Az egyik 11 exonos génklád azonban tartalmazta a varintermelő AutoCBDV genomot és a potenciális varintermelő Durban Poisont, amelyek csökkent funkciójú variánsok lehetnek. Egyes beszámolók szerint a varin kémiai fenotípushoz nem kapcsolódik meghatározott földrajzi eredet [57]. Más vizsgálatok ugyanakkor magas varin kannabinoidszintű növényeket jelentenek Afrika déli régióiból és Ázsia bizonyos térségeiből [52,58].
Együttesen a BKR génfilogénia és a teljesgenom k-mer alapú klaszterezési elemzés ázsiai eredetet valószínűsít az e nemesítési projektben használt varin kannabinoid gének számára. E bioszintetikus útvonalak mélyebb megértése fokozza képességünket a változatos kannabinoid-termelés szelektálására és optimalizálására, és utat jelez a magolaj-lipidprofilok javítása felé.
Következtetések
A 193 kannabiszgenom elemzése feltárta, hogy a globális diverzitás továbbra is alulmintázott, különösen az ázsiai csíraplazma alulreprezentált. Fenotípusos hasonlósága ellenére az európai kenderhez, az ázsiai kender erősen divergáló genomi régiókat hordoz, amelyek közül néhány közelebb áll az észak-amerikai drogtípusú kannabiszhoz; ez feltáratlan vad rokonokra és megoldatlan taxonómiára utal. A TE-aktivitás és hibridizáció – nem pedig teljesgenom-duplikáció – hajtja a kannabiszgenom evolúcióját.
Az SV-k olyan korábban rejtett diverzitást tárnak fel, amelyet a rövidolvasat-szekvenálás elmulasztott. Míg a kannabinoid-szintáz gének korlátozott variációt mutatnak, a zsírsav-anyagcseréhez, növekedéshez, védekezéshez és terpén-bioszintézishez kapcsolódó gének kiterjedt diverzitást és kópiaszám-változatosságot mutatnak.
Teljesen fázisolt kannabisz X és Y kromoszómákat állítottunk össze, változó SDR-PAR határt és egyedi, hím-specifikus homológokat azonosítva a nagy Y kromoszómán; ezek befolyásolhatják a virágzási időt és a fejlődést, új nemesítési célpontokat kínálva.
Végül a zsírsav-bioszintézis gének (például ALT és BKR) kiterjedt változatosságának felfedezése arra utal, hogy a kannabisz kiaknázatlan potenciált hordoz a lipidanyagcserében. A kannabinoid-bioszintézis és a magolaj-útvonalak átfedése miatt a hagyományos észak-európai kendermagolaj-génkészleten túli diverz szülővonalak hibridizálása új lipidprofilokat és tulajdonságokat eredményezhet.
Az ázsiai kender és a vad kannabisz megőrzése, valamint hasznosítása kritikus lesz a kannabisznemesítés és az agronómiai, illetve gyógyszerészeti potenciál fejlesztése szempontjából.
Módszerek
Növényi anyag
A C. sativa pángenom-mintákat több forrásból választottuk ki a genetikai diverzitás, a történeti háttér és az agronómiai érték maximalizálása érdekében. A pángenom nagy része az Oregon CBD (OCBD) nemesítési programból származik, amely elit kultivárokat, az 1970-es évektől napjainkig eredő alapító marihuána-vonalakat, valamint a nemesítési program különböző aspektusaihoz használt elit triókat foglal magában.
A fennmaradó kultivárok az Egyesült Államok Mezőgazdasági Minisztériumának Germplasm Resource Information Network (GRIN) gyűjteményéből, a Német Szövetségi Génbankból (IPK Gatersleben), valamint a Salk Institute különböző nemesítőktől származó gyűjteményeiből erednek. A pángenom európai és ázsiai rost- és magkender, elvadult populációk, észak-amerikai marihuána (I. típus), valamint nagy kannabinoidhozamú észak-amerikai (CBD vagy CBG) kender (III. és IV. típus) mintáit tartalmazza.
További kannabinoid-diverzitást képviselnek a CBD vagy THC pentil- vagy propil- (varin-) homológjainak magas expresszióját mutató kemotípusok, valamint kannabinoidmentes (V. típusú) növények. A virágzási idő változatosságát is lefedtük normál rövidnappalos és nappalhossz-semleges (autoflowering) fenotípusok bevonásával.
EH23 fázisolt, haplotípus-felbontású, kromoszómaléptékű horgonygenom
Az EH23a (HO40) és EH23b (ERB) haplotípus-felbontású összeállítások az ERBxHO40_23 egyedhez tartoznak, amely F1 hibrid az OCBD két saját, nőivarú beltenyésztett szülői vonala, az ERB és a HO40 keresztezéséből. Az ERB DN (autoflower), III. típusú (CBDA-domináns) növény, amely a drogtípusú csoporton belül az európai HC kenderhez közelebb áll. A HO40 I. típusú propil-kannabinoid- (THCVA és THCA-) termelő, rövidnappalra reagáló virágzású növény, és az MJ drogtípusú marihuána csoport része, közelebbi affinitással az ázsiai kenderhez.
A genetikailag nőivarú (XX) ERB növényt ezüst-tioszulfát-kezeléssel hímvirágok termelésére indukálták, majd ezzel porozták a HO40-et. Az F1 populációból egy egyedet (ERBxHO40_23) választottak ki genomszekvenálásra. Áramlási citometriával az ERB × HO40_23 kezdeti diploid genom-méretét 1445,6 Mb-ra (722,8 Mb haploid genom-méret) becsülték. Nagy molekulatömegű (HMW) DNS-t levélszövetből extraháltak.
A DNS-extrakciót és könyvtárkészítést követően HiFi olvasatokat generáltak Pacific Bioscience (PacBio) Sequel II platformon. A Hifiasm v0.16.1-et Hi-C olvasatokkal együtt használták a kezdeti összeállítások előállítására [59]. Az összeállítás után a Hi-C olvasatokat a Hifiasm_HiC kontigokhoz illesztették a Juicer v1.6.2 folyamattal [60], majd a 3D-DNA pipeline 180922 verziójával rendezték és orientálták [61]. Az állványozott összeállításokat ezután Juicebox v1.11.08 segítségével manuálisan javították [62].
EH23 F2 populáció
A fent leírt teljesgenom-szekvenálási adatok mellett az ERBxHO40_23 egyedet ezüst-tioszulfát által indukált, kiválasztott virágok maszkulinizációjával önbeporzással szaporították, hogy F2 térképezési populációt hozzanak létre. E F2 populáció egyedeit autoflower és varin-tartalom alapján pontozták, majd NRGene (Nrgene Technologies) Illumina 100 bp olvasatokkal szekvenálta. Illumina WGS genotipizálási futásokat végeztek e populáció 288 növényén, valamint az ERBxHO40_23 szülőn.
A szekvenciákat Trim_galore segítségével vágták (–2 colour 20), aminek eredményeként 271 egyed maradt elemzésre [63]. A minták átlagos lefedettsége 8,5× volt. A mintákat Minimap segítségével illesztették az EH23b.softmasked.fasta referenciához. A variánsokat Freebayes-szel hívták (-g 4500 -0 -n 4 –trim-complex-tail –min-alternate-count 3) [64]. A Bcftools segítségével QUAL > 20 pontszámra szűrtek (99% valószínűség, hogy a variáns létezik) [65].
Végül a Vcftools eszközöket alkalmazták további SNP-szűrésre: –remove-indels –minGQ 20 –maf 0.25 –max-missing 1 –min-alleles 2 –max-alleles 2 –stdout –recode [66]. Csak azokat a helyeket tartották meg, amelyek az ERBxHO40_23 mintában heterozigótaként (0/1) lettek pontozva; ez 93 251 SNP-t eredményezett.
EH23 F2 kannabinoid HPLC-módszerek
A nagyhatékonyságú folyadékkromatográfiát (HPLC) korábban részletesen leírt protokoll szerint végezték [67], hogy meghatározzák a propil- és pentil-kannabinoidok relatív tartalmát a vizsgálatban használt minden növényben, beleértve az F2 utódokat is. Röviden: minden egyedtől érett virágszövetet gyűjtöttek, -80 °C-on lefagyasztották és homogenizálták, majd a kannabinoidokat metanolban extrahálták.
EH23 RNS-szekvenálás
Az ERBxH040-21 palántákat kontrollált környezeti körülmények között nevelték. A növények fejlődése során különböző szöveteket gyűjtöttek, beleértve a korai és késői virágokat, leveleket, 12 órás induktív fényrendszerben nevelt leveleket, gyökereket és hajtáscsúcsokat. A teljes RNS extrakcióját a QIAGEN RNeasy Plus Kit segítségével, a gyártói protokoll szerint végezték. A teljes RNS-t Qubit RNA Assay-vel és TapeStation 4200-zal kvantifikálták.
A könyvtárkészítés előtt DNáz-kezelést, majd AMPure gyöngyös tisztítást és QIAGEN FastSelect HMR rRNS-depléciót végeztek. A könyvtárkészítés a NEBNext Ultra II RNA Library Prep Kit segítségével, a gyártói protokoll szerint történt. A könyvtárakat NovaSeq6000 platformon futtatták 2 × 150 bp konfigurációban.
EH23 haplotípus-expressziós elemzés
A génexpressziós szinteket Salmon v1.6.0-val mérték [68]. Röviden, a szekvenálás nyers páros végű rövid olvasatait mindkét haplotípus (EH23a és EH23b) CDS-eihez térképezték, és a mennyiséget transzkript/millió (TPM) értékben becsülték a további elemzéshez. A leképezési arányokat samtools flagstat segítségével számították [65]. Egy adott gén minimális TPM-küszöbe ≥0,1 volt.
A haplotípus génpárokat reciprok legjobb találatok és szinténia alapján azonosították blastp és MCScanX segítségével [69], és csak mindkét haplotípusban megosztott géneket vontak be. Legalább 95%-os szekvenciahasonlóságot és 5 TPM különbségi küszöböt írtak elő a haplotípusok között. A vizualizáció Matplotlib [70], SciPy [71] és NumPy [72] kombinációjával készült; az expressziós értékeket hőtérképeken log2TPM-ként mutatták be a log fold change ábrázolására.
A biológiai folyamat GO-kifejezések gazdagodását topGO-val [73] végezték a következő paraméterekkel: resultWeight <- runTest(topGOdata, algorithm = „weight01”, statistic = „fisher”). Többszörös tesztkorrekciót alkalmaztak: fullResults$p.adj <- p.adjust(as.numeric(fullResults$weightFisher), method = „fdr”). A háttér génuniverzum minden olyan gént tartalmazott, amely EH23a vagy EH23b alapján GO-kifejezéssel rendelkezett.
Ace High ivar szerint torzított génexpressziós elemzés
Négy Ace High növényből – két hímből és két nőivarúból – ugyanazon fejlődési időpontban, 08:00 és 20:00 órakor gyűjtöttek virág- és levélszövetet, összesen 16 mintát. Mivel az Ace High hímek normál kültéri körülmények között több héttel a nőivarú növények előtt virágoznak, a növényeket hosszú nappalon csíráztatták és nevelték, majd induktív rövidnappalos körülmények közé helyezték virágzásra; ez azt eredményezte, hogy a hím és nőivarú növények egy időben fejlesztettek virágokat.
A mintákat a nap két időpontjában gyűjtötték, hogy a cirkadián vagy diurnális expressziótól függetlenül minden transzkriptumot lefedjenek [74]. Az RNS-t Qiagen Plant RNA kittel extrahálták. A könyvtárkészítés Oxford Nanopore Technologies (ONT) teljes hosszúságú cDNS kittel történt. A teljes hosszúságú cDNS-t minimap2 (v2.24) [75] segítségével illesztették a haplotípus-felbontású Ace High (AH3Ma/b) genomokhoz, és az expressziót Salmon v1.6.0-val mérték [68].
Ivar szerint torzított expressziót minden szövetspecifikus hím és női mintára hozzárendeltek. Minden ivarspecifikus szövet négy ismétléssel rendelkezett. Két torzított expressziós kategóriát határoztak meg: az egyikben az átlagos expresszió legalább 5 TPM-mel magasabb volt az egyik ivar mintáiban a másikhoz viszonyítva; a másikban csak hím- vagy csak női expressziót mutató géneket definiáltak, ahol a gén az egyik ivarban nem expresszálódott (minden ismétlésben 0,0 TPM), a másikban pedig átlagosan legalább 1,0 TPM volt. A topGO-val végzett GO-elemzéshez a két kategóriát összevonták.
A teljesen szinténikus géneket a négy X és Y kromoszómát tartalmazó genom (AH3Ma/b, BCMa/b, GRMa/b és KOMPa/b) halmazában genespace segítségével azonosították, és PAR, SDR vagy X-specifikus régió szerinti helyzetük alapján csoportosították.
Hi-C könyvtárkészítés és szekvenálás
A Dovetail Omni-C könyvtárhoz a kromatint formaldehiddel rögzítették a sejtmagban, majd extrahálták. A rögzített kromatint DNase I-gyel emésztették, a kromatinvégeket javították és biotinilált hídadapterhez ligálták, majd az adaptert tartalmazó végeket közelségi ligálással kapcsolták. A közelségi ligálás után a keresztkötéseket visszafordították, a DNS-t tisztították, majd a biotint nem belsőleg hordozó fragmentumok eltávolítására kezelték.
A szekvenálási könyvtárakat NEBNext Ultra enzimekkel és Illumina-kompatibilis adapterekkel készítették. A biotint tartalmazó fragmentumokat streptavidin gyöngyökkel izolálták a PCR-dúsítás előtt. A könyvtárat Illumina HiSeqX platformon szekvenálták, körülbelül 30× lefedettséget eredményezve. Ezt követően a HiRise MQ > 50 olvasatokat használt állványozáshoz. További Hi-C könyvtárak Phase Genomics Proximo Hi-C Kit (Plant) version 4 segítségével készültek.
HMW DNS izolálás és genomszekvenálás
Minden mintát PacBio Sequel II platformon szekvenáltak. A „Michael” forrásból származó minták esetében HMW DNS-t Carlson Lysis pufferrel és Qiagen Genomic tippekkel izoláltak az ONT „Plant leaf gDNA” Arabidopsis protokoll szerint. A DNS-t tovább méret-szelektálták 10-25 kb-nál hosszabb fragmentumokra ONT Short Fragment Eliminator Kit (EXP-SFE001) segítségével. A HMW DNS-t Tapestation Genomic DNA ScreenTape vagy Femto Pulse Genomic DNA 165 kb Kit segítségével ellenőrizték.
Az „OCBD” forrásból származó mintáknál módosított protokollt alkalmaztak [76]. Röviden: a mintákat folyékony nitrogénben, mozsárban őrölték; két kloroform:izoamil mosási ciklust végeztek, és az eredeti protokoll helyett Total Pure NGS gyöngyöket (Omega Biotek) használtak. A genom-DNS minőségét és tisztaságát NanoDrop One eszközzel értékelték a könyvtárkészítés előtt.
A folyamatos hosszú olvasatú (CLR) könyvtárak a PacBio PN 101-693-800 V1 protokoll szerint készültek. A genom-DNS méretválasztása Blue Pippin U1 High Pass 30-40 kb kazettával történt, 30-40 kb kezdő küszöbbel, 60-90 kb fragmentumeloszlások előállítására. A HiFi circular consensus sequencing (CCS) könyvtárak a PacBio PN 101-853-100 V5 protokoll szerint készültek. A körülbelül 18 kb modális csúcsú, nyírt gDNS-fragmentumeloszlásokat Covaris g-Tube eszközökkel és Blue Pippin S1 High Pass 6-10 kb kazettákkal hozták létre, eltávolítva minden 10 kb alatti fragmentumot.
Pángenom-összeállítás és állványozás
Minden Hifiasm_HiC, Hifiasm_Trio_RagTag, Hifiasm_RagTag és Hifiasm jelölésű genomot Hifiasm v0.16.1-gyel állítottak össze [59]. Ha rendelkezésre álltak, Hi-C adatok és HiFi szülői trio adatok is bekerültek az összeállítási folyamatba, meghatározva a Hifiasm_HiC és Hifiasm_Trio_RagTag típusokat. A CLR összeállítások PacBio SMRT Tools 9.0 Suite-ból származó FALCON Unzip [77] segítségével, a CCS jelölésű genomok pedig HiCanu v2.2-vel készültek [78].
Az összeállítást követően a Hi-C olvasatokat Juicer v1.6.2 [60] segítségével illesztették a Hifiasm_HiC kontigokhoz, majd a 3D-DNA pipeline 180922 verziójával rendezték és orientálták [61]. Az állványozott összeállításokat Juicebox v1.11.08 [62] segítségével manuálisan javították. A Hifiasm_RagTag és Hifiasm_Trio_RagTag összeállításokat a 24 Hi-C állványozott genom feldarabolt kromoszómái segítségével állványozták, és yak-0.1 eszközzel ellenőrizték.
A Sourmash v4.6.1 [79] Jaccard-hasonlósági mátrixot készített a kromoszómák és minden nem állványozott összeállítás között; az 1-től X-ig terjedő kromoszómák leginkább hasonló verzióit konkatenálták, hogy RagTag v2.1.0 [80] segítségével állványozási referenciát alkossanak. Ha a hasonlósági mátrix az Y kromoszómát azonosította legjobb egyezésként, az összeállítás állványozatlan maradt. Az összes összeállításon BUSCO v5.4.3 [79] eudicots_odb10 adatkészlettel és assembly-stats v1.0.1 eszközzel mérték a teljességet és kontinuitást.
Referenciaalapú gráfépitás Minigraph-Cactus-szal
A 78 állványozott és softmaskolt összeállítás gráf-pángenomját Minigraph-Cactus [20] segítségével állították elő. A cactus-pangenome parancsot Apptainer (v1.1.8) Image [81] környezetben használták, a következő paraméterekkel: –reference EH23a EH23b –vcf –vcfReference EH23a EH23b –giraffe –chrom-og –chrom-vg –viz –gfa –gbz. A seqFile bemenet és a különböző formátumú (vg, paf, hal stb.) kimeneti gráfok a https://resources.michael.salk.edu címen találhatók.
A pángenom variánsait minden összeállítás koordinátái szerint is összeállították: vg deconstruct -a -C (vg tools v1.61.0 „Plodio”) segítségével vcf fájlokat származtattak a Minigraph-Cactus gfa kimenetből, majd vcfbub –max-ref-length 100000 –max-level 0 használatával lapították a beágyazott variánsokat és eltávolították a 100 kb-nál hosszabbakat [20,82,83].
Referenciafüggetlen gráfépitás PGGB-vel
Beviteli szekvenciák és orientáció
Két verziót készítettek minden PGGB gráfból: egyet az „Assembly files” táblázatban és a JBrowse példányban közölt fasta fájlokkal (kevert orientáció), és egyet olyan fasta fájlokkal, amelyek szekvenciáit az EH23a megfelelő homológ kromoszómájának plusz szálához igazodó, konzisztens orientációra állították.
A 16csatAsms PGGB gráfhoz minden autoszomális kromoszómára egy gráfot készítettek 16 állványozott és softmaskolt összeállításból: AH3Ma, AH3Mb, BCMa, BCMb, EH23a, EH23b, GRMa, GRMb, KCDv1a, KCDv1b, KOMPa, KOMPb, MM3v1a, SAN2a, SAN2b és YMv2a. Kromoszómánként egy kombinált fasta fájl szolgált bemenetként a PGGB számára. A teljes, összesített gráf helyett kromoszómánkénti gráfokat építettek a genomok mérete és repetitív tartalma miatti számítási igények csökkentésére.
A 13csatSexChroms PGGB gráfhoz a 13 állványozott és softmaskolt ivari kromoszóma-szekvenciát egyetlen fasta fájlba kombinálták: AH3Ma.chrX, AH3Mb.chrY, BCMa.chrX, BCMb.chrY, EH23a.chrX, GRMa.chrY, GRMb.chrX, KCDv1a.chrX, KCDv1b.chrX, KOMPa.chrX, KOMPb.chrY, SAN2a.chrX és SAN2b.chrX.
A gráfgeneráláshoz Nextflow v24.04.3.591684-et használtak az nf-core/pangenome v1.1.2 – canguro deployment [85,86] PGGB [22] futtatására, nextflow singularity profilban. Az alapértelmezett PGGB-beállításokat használták. A 13csatSexChroms gráfnál a –vcf_spec jelzőt alkalmazták, majd a vcfbub-bal lapították a beágyazott variánsokat és eltávolították a 100 kb-nál hosszabbakat. A 16csatAsms gráfnál ehelyett vg deconstruct -a segítségével állították össze a variációt a végső gfa fájlokból, autoszómánként; a vcf fájlokat bcftools-szal konkatenálták, majd vcfbub-bal szűrték.
Vizualizáció és rövidolvasatok térképezése gráf-pángenomra
A gráf-pángenomok vizualizációi a PGGB-pipeline konzisztens orientációjú bemenetekkel futtatott FINAL_GFA fájljaiból készültek. A gfa fájlokból vg convert [82,83] segítségével vg fájlokat származtattak. A prepare_vg.sh és prepare_chunks.sh szkriptek segítségével a pángenom-variáció érdekes régióit a Sequence Tube Map szerver helyi példányában vizualizálták.
Az EH23 F2 populáció és Ren et al. [2] rövid olvasatait vg giraffe segítségével illesztették a pángenomgráfhoz [87]. Az összegző statisztikákat vg stats [82] gyűjtötte; a GAM fájlok olvasattámogatását vg pack számította, és az F2 térképezési populáció variánsait vg call [88] hívta. A VCF fájlok további feldolgozása BCFtools [65] és VCFtools [66] segítségével történt, a lineáris referencián alapuló VCF-fel való összehasonlítás érdekében.
Gráf-pángenom adatelérhetőség és metilált citozinok bázishívása
A fent leírt gráf-pángenomok bemeneti és kimeneti fájljai – a Minigraph-Cactus által generált 78csatHaps, valamint a PGGB által generált 16csatAsms és 13csatSexChroms – a https://resources.michael.salk.edu címen érhetők el. A VCF fájlokat sávként hozzáadták a Cannabis genomes JBrowse példányához ugyanott.
A nyers ONT FAST5 fájlokból származó genomi olvasatokat metilációhívásra használták. Az ugyanazon egyedekből generált genomösszeállítások referenciaként szolgáltak az illesztéshez. A FAST5 adatokat a pod5 csomaggal POD5 formátumra konvertálták. A metilációhívást az ONT Dorado 0.3.4 bázishívó szoftverével végezték. A Dorado a nyers POD5 adatokat és a referenciát használja metilált citozinok azonosítására, a szekvenálási feltételekhez illeszkedő, R9.4.1 vagy R10.4.1 pórustípusra és 400 bps transzlokációs sebességre tanított, szuper nagy pontosságú (SUP) modellel.
Minden mintához az összeállított genomok szolgáltak referenciaként az 5mC és 5hmC metilációhívásokat tartalmazó MM/ML tag-es BAM fájlok előállításához. Ezeket modkit segítségével pileupolták, és az összevont 5mC + 5hmC hívásokat az összes CG-helyen genomszintű metilációs gyakoriságok számítására használták.
Gén- és ismétlődés-előrejelzés
A génmodellek előrejelzése több lépésből álló pipeline-nal történt, és minden összeállításra alkalmazták. Először RepeatModeler [89] segítségével kurált ismétlődési könyvtárat hoztak létre kis számú, kiváló minőségű Cannabis összeállításból és meglévő repeat-könyvtárakból. Az ismétléseket OrthoFinder (v2.5.4) [90] segítségével csoportosították deduplikáláshoz. A végső ismétlődési könyvtár minden repeat-ortocsoportból a szekvenciák 10%-át tartalmazta (minimum 1 szekvencia), összesen 6262 szekvenciával 5793 csoportból.
Az ismétléskönyvtár létrehozásához használt források: Finola (GCA_003417725.2), CBDRx (GCF_900626175.2), Purple_Kush (GCA_000230575.5), ERBxHO40_23, I3, JL (GCA_013030365.1), ERB_F3, Cannbio-2 (GCA_016165845.1), W103, JL_Mother (GCA_012923435.1), FB30, TS1_3_v1 és HO40. Mind a 193 genomon RepeatMasker (v4.1.2) [91] segítségével maszkolták az ismétlődéseket.
A génmodelleket TSEBRA pipeline-nal (Braker v2.1.6) [92] jelezték előre. A TSEBRA futtatásához Snakemake munkafolyamatot fejlesztettek, amely elérhető: https://gitlab.com/salk-tm/snake_tsebra. Bizonyítékként több meglévő fehérjekönyvtárat használtak kannabiszból és más szervezetekből, köztük Arabidopsis thaliana, Theobroma cacao, Glycine max, Rhamnella rubrinervis, Ziziphus jujuba, Trema orientale, Vitis vinifera, Prunus persica, Morus notabilis, C. sativa és H. lupulus fajokból.
Az RNS-seq könyvtárakat hisat2 (v2.2.1) [93] vagy minimap2 (v2.24) [75] segítségével illesztették, attól függően, hogy rövid olvasatokról vagy teljes hosszúságú cDNS-ről volt szó. A rövidolvasatú Illumina adatokat fastp [94] vágta. Az expressziós adatokat génmodell-bizonyítékként építették be a TSEBRA pipeline-ba. A génmodellek feltételezett funkcionális annotációit eggnog-mapper (v2.0.1) [95] rendelte hozzá.
A génmodell-minőséget és teljességet úgy értékelték, hogy a genom BUSCO (v5.4.3) [96] pontszámokat a proteom BUSCO pontszámokkal hasonlították össze az eudicots_ocdb10 adatkészleten. Az EDTA v1.9.6 [97] szintén használatban volt a kannabisz-pángenom TE-inek azonosítására az EDTA.pl –genome {inputFastaFile} –anno 1 –threads 32 paranccsal.
Ideogrammódszerek
A 78 kromoszómaszintű, haplotípus-fázisolt genom minden kromoszómapárjához ideogramokat készítettek R-ben ggplot2 segítségével. A kromoszómák hosszát a nuccomp.py határozta meg, és ggplot::geom_rect() segítségével indították az ábrát. Minden kromoszómára egymillió bázispáros ablakokat hoztak létre; ezekben a CpG motívumok számát motif_counter.py számolta. A CpG-számot az ablakmérettel osztva rátává alakították, figyelembe véve az utolsó, egymillió bázispárnál rövidebb ablakokat is.
A rátákat kromoszómánként skálázták a minimum levonásával és a maximum szerinti osztással. A centromerikus régió CpG-motívum-gazdagodásának vizuális hangsúlyozására a CpG-rátából inverzet képeztek. Ezt a skálázott, inverz CpG-rátát használták minden 1 Mb-os ablak szélességéhez, a színezés pedig génsűrűség alapján, viridis magma palettával történt.
A kromoszómapárok közötti strukturális variációt minimap2 [75] illesztésekkel határozták meg, az összehasonlításokat SyRI [98] annotálta. A szinténikus és invertált régiókat ggplot2::geom_polygon() segítségével rajzolták meg, a plotsr [99] által inspirált, R-ben megvalósított módon. Az EH23 A és B haplotípusaiban a jelölt lókuszok helyét BLASTN [100] határozta meg a CBCA, CBDA, THCA szintáz és olivetolsav-cikláz lekérdezési szekvenciák alapján; ezeket centromerikus, telomerikus és rRNS szekvenciákkal kombinálták, majd az eredményeket ideogramokra vitték fel.
Centromer- és telomeranalízis
Az ONT- és PacBio-alapú hosszúolvasatú genomösszeállítások lehetővé teszik egyes erősen repetitív centromer- és telomerszekvenciák összeállítását [101]. A centromereket a genomok tandem repeat finder (TRF; v4.09) általi keresésével, módosított beállításokkal (1 1 2 80 5 200 2000 -d -h) azonosították [102]. A tandem ismétléseket újraformázták, összegezték és ábrázolták, hogy a korábbi módszerek szerint a legmagasabb kópiaszámú tandem ismétlést megtalálják a centromerek azonosítására.
A telomereket két módszerrel becsülték. Először a TRF kimenetet olyan 7 periódusú ismétlésekre kérdezték le, amelyek a kanonikus telomer bázisismétlés 14 különböző verziójának feleltek meg (AAACCCT, AACCCTA, ACCCTAA, CCCTAAA, CCTAAAC, CTAAACC, TAAACCC, TTTAGGG, TTAGGGT, TAGGGTT, AGGGTTT, GGGTTTA, GGTTTAG és GTTTAGG). Másodszor a nyers ONT és PacBio olvasatokban telomerszekvenciákat kerestek saját TeloNum algoritmussal [103].
Bár az eredmények pángenom-összeállításonként változtak, általánosságban a kromoszóma végén telomerszekvencia volt található, PacBio összeállításoknál átlagosan 16 kb, ONT összeállításoknál 60 kb hosszal. Az ONT és PacBio telomerhossz közötti különbségek valószínűleg az input olvasathosszakat tükrözték (>100 kb, illetve 15-20 kb). A nyers olvasatok TeloNum-elemzése alátámasztotta az összeállítások eloszlásait: a legtöbb kromoszómán volt telomerszekvencia, de az valószínűleg rövidebb volt a ténylegesnél. A kannabisz telomerjei az eudikóták között a hosszabbak közé tartoznak, amit a gyógyászati felhasználásokra jellemző klonális szaporítás magyarázhat [104].
A centromerszekvenciát az alapján a hipotézis alapján azonosították, hogy ez lesz a genomok legbőségesebb ismétlése, amely magasabb rendű ismétléses (HOR) szerkezettel is rendelkezik [101,105]. A PacBio HiFiasm összeállításokban két HOR-os ismétlést azonosítottak, míg az ONT-összeállításokban és a korábbi ONT-alapú CBDRx összeállításban csak egyet [11]. A legmagasabb kópiaszámú ismétlés 370 bp volt, amely 20-30 Mb között változott, 740 és 1110 bp-os HOR-ral. A második legmagasabb és az ONT-összeállításokban egyetlenként talált ismétlés 237 bp volt, 3-5 Mb változással és 474, illetve 711 bp-os HOR-ral.
A 370 bp-os ismétlés kromoszómafelbontású genomokra történő leképezése azt mutatta, hogy ez elsősorban a kromoszómák végén, a telomerszekvencia mellett helyezkedik el, ami arra utalt, hogy a CS-1 szubtelomerikus ismétléssel lehet rokon [106]. A feltételezett 370 bp-os centromerikus ismétlés és a CS-1 szubtelomerikus ismétlés összehasonlítása azt mutatta, hogy ugyanarról az ismétlőelemről van szó. Ezzel szemben a feltételezett 237 bp-os centromerikus ismétlés főként a 6. és 8. kromoszóma előre jelzett centromerrégiójában fordult elő, bár kisebb tömbök minden kromoszómán megtalálhatók voltak.
Riboszomális DNS detektálása és kvantifikálása
A riboszomális DNS (rDNS) 45S (18S, 5.8S és 26S), valamint 5S szekvenciákat a CBDRx/CS10 összeállításban azonosították, és BLAST-tal vetették össze a pángenom-összeállításokkal. Az állványozott genomokban a 45S tömb döntően a 8. kromoszóma akrocentrikus végén helyezkedett el, míg az 5S kizárólag a 7. kromoszómán, a kannabinoid-szintáz-kazetta tömbje között fordult elő, összhangban a fluoreszcens in situ hibridizációval közölt eredményekkel [106].
Részleges tömböket ugyanakkor egyes összeállításokban minden kromoszómán találtak. A különböző kromoszómákon található részleges tömbök eloszlása a genomok közötti változatosságot tükrözheti, mivel némelyik hasonló helyeken fordul elő az összeállítások között. A legtöbb tömb nem állványozott kontigokon található, ami arra utal, hogy a különböző kromoszómákon megjelenő variábilis tömbök félre-összeállításokból eredhetnek. Általánosságban a kannabiszgenomban átlagosan 1000 45S és 2000 5S tömb található; egyes összeállításokban az 5S tömb teljesen összeállt a 7. kromoszómán.
Allélgyakorisági módszerek és PanKmer-genomelemzés
A VCF formátumú genotípusadatokat vcfR [108] segítségével vitték be R-be [107]. Az allél- és heterozigóta-számlálást vcfR-rel végezték. Wright FIS értékét [109] a véletlenszerű Hardy-Weinberg várakozástól való heterozigozitási eltérés mérésére számították: FIS = (HS – HO) / HS, ahol HO a megfigyelt heterozigóták száma arányosan, HS pedig az allélgyakoriságokból számított várható heterozigóta-gyakoriság. A szórásdiagramok ggplot2-vel, a grafikus panelek ggpubr segítségével készültek.
PanKmerrel két 31-mer indexet készítettek: egy „teljes” indexet 193 Cannabis-összeállításból és egy „csak állványozott” indexet 78 állványozott összeállításból, alapértelmezett pankmer index paraméterekkel. A páronkénti Jaccard-hasonlóságokat pankmer adj-matrix és pankmer clustermap –metric jaccard segítségével számították és ábrázolták. A gyűjtőgörbéket mindkét indexre pankmer collect paranccsal, alapértelmezett paraméterekkel számították; az elemzés szkriptjei GitHubon érhetők el.
Génalapú pángenom elemzése és gyűjtőgörbék
A génalapú pángenomot minden olyan géncsalád (ortocsoport) halmazaként definiálták, amely a pángenom legalább egy genomjában képviselt. Mind a 193 C. sativa genomhoz – külön halmazként a 78 kromoszómaszintű, haplotípus-fázisolt genomhoz is – minden nagy megbízhatóságú génelőrejelzés elsődleges transzkriptjét választották reprezentánsnak. Az elsődleges transzkripteknek megfelelő fehérjéket Orthofinder (v2.5.4) segítségével ortocsoportokba klaszterezték [90].
Az elsődleges transzkript CDS-eket egyetlen FASTA fájlba egyesítették, és az egzakt duplikátumokat SeqKit (2.7.0) távolította el [110]. A feltételezett kontaminánsokat olyan kontigokon előre jelzett transzkriptek alapján azonosították és távolították el, ahol az előrejelzések kevesebb mint 90%-át annotálta eggNOG-mapper [95] viridiplantae vagy eukaryote kategóriába. Az annotálatlan gének problémájának mérséklésére minden elsődleges transzkript CDS-ét minimap2-vel [75] illesztették mind a 193 (illetve 78) kannabiszgenomhoz, splice beállításokkal.
Egy adott genomban, ha egy illesztett CDS-szekvencia legalább 60-as leképezési minőségű volt, CIGAR egyezéseinek száma legalább a lekérdezési hossz 80%-át elérte, és nem fedett át közvetlenül annotált gént, akkor annotálatlan génnek tekintették, és ortocsoportját jelenlévőként jelölték az adott célgenomban. Azok az ortocsoportok, amelyeknek legalább egy képviselője minden genomban jelen volt, a maggenomhoz tartoztak; a többi a változó genomhoz. Az ortocsoportok jelenlétét vagy hiányát táblázatban rögzítették.
A pángenomikában a gyűjtőgörbék (pángenom-ritkítás) a haplotípusok száma (H) és a géncsaládok/ortocsoportok száma (X) közötti kapcsolatot mutatják. Az ortocsoportok pontszáma azt jelenti, hány haplotípusban van jelen az adott ortocsoport. A pángenom-gyűjtőgörbe C(h) a H teljes halmazból véletlenszerűen kiválasztott h haplotípus részhalmazban várható ortocsoportszámot adja. A maggenom-gyűjtőgörbe ennek megfelelően a h haplotípusban közösen jelen lévő ortocsoportok várható számát becsli. A képletek a hipergeometrikus túlélési függvényre, illetve a hipergeometrikus kumulatív eloszlásfüggvény 1-ből való kivonására épültek. A k-mer alapú gyűjtőgörbék ugyanilyen módon készültek, csak az egység ortocsoport helyett k-mer volt.
K-mer elemzés és mag/dispensable gének azonosítása
Ren et al. [2] Illumina rövidolvasat-szekvenciáit Trim_galore segítségével vágták, majd alacsony bőségű olvasatokra szűrték, és sourmash sketch dna parancs segítségével 31-mer vázlatot készítettek. Minden pángenom-összeállítást 31-mer gyakoriságokra is elemeztek. A páronkénti Illumina-olvasat és pángenom-összeállítás mintákat sourmash compare paranccsal hasonlították össze, majd a 31-mer távolságokat R-ben, hclust átlagos módszerrel ábrázolták.
A mag és dispensable (majdnem mag, felhő, héj, privát) géneket ortocsoport-tagság alapján rendelték hozzá. A maggéneket a genomok 100%-ában (193 genom), a majdnem mag géneket 95-99%-ban (183-192 genom), a héjgéneket 5-94%-ban (10-182 genom), a felhőgéneket 2-5%-ban (3-9 genom), az egyedi géneket pedig 0,5-1%-ban (1-2 genom) jelen lévőként definiálták [111]. Az elemzést mind a 193 genomon elvégezték, és populáció szerint is vizualizálták.
A 103 kontigszintű összeállításnál csak az EH23a tíz kromoszómájához hasonló kontigokat vették figyelembe. A génkészleteket úgy szűrték, hogy csak a tíz kromoszómán és homológ kontigokon található géneket tartalmazzák. A mag, héj, felhő, majdnem mag és egyedi géncsoportokra topGO-val funkcionális gazdagodási elemzést végeztek, ahol a háttér az adott genom minden GO-kifejezéssel rendelkező génje volt.
Ismétlődéselemzés
A TE-k divergenciaidejét a T = (1 – identity) / 2µ egyenlettel becsülték, ahol az identity az EDTA GFF3 kimenetből származott [97]. A szubsztitúciós rátaként Arabidopsis alapján 6,1 × 10^-9 értéket használtak [112,113]. Az elemzést minden genomon elvégezték.
A solo LTR-ek és intakt LTR-RT-k azonosításához az EDTA pipeline-t alkalmazták 193 kannabiszgenomon [97]. Solo LTR-eket úgy azonosítottak, hogy először begyűjtötték azokat az LTR-eket, amelyeket nem soroltak intakt LTR-RT-k közé, majd küszöböket alkalmaztak a solo LTR-ek elkülönítésére a csonka és intakt LTR-ektől, valamint az LTR-RT belső szekvenciáitól. A küszöbök közé tartozott a minimum 100 bp hossz, 0,8 azonosság a referencia-LTR-hez, minimum 300 illesztési pontszám, azonos LTR-RT ID-vel rendelkező szomszédos annotációk kizárása, valamint legalább 5000 bp távolság a legközelebbi solo-LTR, intakt LTR vagy belső szekvencia felé.
A genomi jellemzőket szegélyező TE-k gazdagodását a PlanTEnrichment részeként bemutatott módszer adaptálásával értékelték. A cél az volt, hogy azonosítsák azokat a TE-ket, amelyek szignifikánsan társulnak egy adott genomi jellemző kategóriához, például kannabinoid-szintáz génekhez. Az enrichment score képlete ES = (a/b)/(c/d), a P-értéket faktoriális kifejezéssel számították, majd statsmodels Python-könyvtárral többszörös tesztkorrekciót végeztek [119,120]. A szignifikanciaküszöb FDR < 0,05 és ES ≥ 2 volt.
A TE-k és gének távolságát bedtools sort és bedops closest-features segítségével számították [122]. A különböző TE-kategóriákhoz társuló gének GO-gazdagodását topGO-val végezték. A kannabinoid-szintázokat körülvevő TE-k filogéniájához a 78 állványozott összeállításban a CBCAS, CBDAS és THCAS gének 2 kb-os upstream és downstream régióit gyűjtötték be, majd a TE-ket bedtools intersect segítségével vonták ki; a fákat IQ-TREE-vel vagy kapcsolódó eszközökkel becsülték és FigTree-ben vizualizálták.
Az EH23 aktív TE-expressziójának elemzéséhez nem redundáns TE-könyvtárat használtak, az olvasatokat a TE-könyvtárra térképezték, és az expressziót TPM-ben mérték. A megfigyelt/várt CpG arányt CpG-sziget definíció alapján értelmezték: >200 bp metilálatlan régió, >50% GC-tartalom és >0,6 megfigyelt/várt CpG arány. A citozinmetiláció idővel CpG dinukleotid-vesztéshez vezethet; e mintázatokat a 2h,k. ábrákon vizualizálták.
SV-ket közvetlenül szegélyező TE-k és szinténiaelemzések
Az SV-altípusok – inverziók, duplikációk, transzlokációk és invertált transzlokációk – minden töréspontja körül 500 bp upstream és downstream régiót (összesen 1 kb) vizsgáltak TE-tartalomra, intakt és fragmentált annotációk alapján. A 78 állványozott, kromoszómaszintű genomot populáció szerint csoportosítva vonták be. Összehasonlításként azonos hosszúságú véletlen ablakokat vettek ugyanabból a genomból és kromoszómából bedtools shuffle segítségével. Csak azokat az eseteket értékelték tovább bedtools intersecttel, ahol egy adott TE-típus egyetlen SV mindkét töréspontjához társult. A statisztikai szignifikanciát SciPy-ben Welch-féle kétoldali t-próbával értékelték.
Az Orthofinder és szinténiaelemzésekhez Orthofinder version 2.5.4-et futtattak a pángenom-összeállításokon és nem kannabisz outgroup genomokon, hogy ortológ csoportokat hozzanak létre. A szekvenciaentrópiát DNS- és fehérjealapú ortocsoportokra számították, többes illesztések oszlopaira. A szinténia vizualizálásához és elemzéséhez genespace version 0.9.3-at használtak R 4.2.2-ben.
Az SV-elemzésben a 78 teljesen állványozott összeállítás haplotípusait az EH23a haplotípus-összeállításhoz illesztették, és Syri segítségével annotálták. A SNP-ket ugyanazon összeállításokból és illesztésekből hívták Syri-vel; a két haplotípus SNP-jeit mintánként fázisolt genotípusként egyesítették, eltávolították az ALT-ként N-t tartalmazó helyeket, majd vcftools segítségével minőségszűrték és ritkították legalább 1000 bp távolságra. Az LD-számításokat plinkkel, majd ld_decay.py segítségével végezték; külön LD-hőtérképeket vcftools és LDheatmap segítségével készítettek.
Szelekció, TreeMix, lokális PCA és rezisztenciagén-analógok
A GO-gazdagodási teszteket R-ben a topGO csomaggal végezték, EH23a nagy megbízhatóságú génannotációit null-eloszlásként használva és klasszikus Fisher-próbával [73]. Az Fst értékeket vcftools segítségével számították minden fázisolt SNP-re és az állványozott összeállítások MJ és kender populációs hozzárendeléseire; a szignifikanciát az értékek felső 5%-a alapján állapították meg. A szelektív söprések XP-CLR modelljét ugyanezekre az SNP-kre és 20 kb-os genomi ablakokra alkalmazták.
A TreeMix modellt csak génmodelleken kívüli SNP-kkel futtatták, -seed 69696969 -m 5 -k 50 -noss -root asian_hemp paraméterekkel. Egytől tízig terjedő migrációs forgatókönyveket szimuláltak, és a log-likelihood alapján rangsoroltak; a végső legvalószínűbb migrációszám öt volt. A lokális PCA-módszert 1000 bp minimális SNP-távolsággal és 100 SNP-s genomi ablakokkal alkalmazták [134].
A növényi betegségrezisztencia-gén analógokat a kódolt fehérjékben található erősen konzervált aminosavmotívumok alapján definiálják. A Drago2 [135] a 78 kromoszómaszintű, haplotípus-felbontású genomban azonosította a növényi rezisztenciagén-analógok között konzervált motívumokat. A nukleotidkötő hely (NBS) és leucindús ismétlés (LRR) doméneket tartalmazó gének készleteit MEME-be vitték a motívumok aminosav-összetételének összehasonlítására.
A lisztharmat-rezisztenciához kapcsolódó gének azonosításához egy CBDRx-ben a 2. kromoszómára térképezett marker szekvenciáját BLASTN lekérdezésként használták az EH23a horgonygenom ellen [136]. A találat egy 46 génből álló klaszterben helyezkedett el, több kinázdoménnel, receptor-szerű kinázzal, NBS/transzmembrán doménnel és coiled-coil kombinációkkal. A top találatok nem fedtek át annotált génekkel, de több genomban nagy azonosságú találat volt jelen; a régiókat kivonták, illesztették a CBDRx génszekvenciához, és maximum-likelihood fát készítettek.
A coiled-coil NBS-LRR gének (CNL-ek) jellegzetes mintázatot mutattak a 3. és 6. kromoszómán. Egyes régiókban 1-5 CNL gén fordult elő, több kivétellel és áthelyezett centromerikus pozíciókkal; ezek a pángenomon belüli jelentős rezisztenciagén-diverzitást jelzik.
Terpén- és kannabinoid-bioszintézis gének azonosítása
Minden Cannabis proteomot 40 926 UniProt fehérjeszekvenciából álló halmazhoz illesztettek blastp segítségével (Embryophyta, reviewed; hozzáférés: 2022. szeptember 20.) [137,138]. A terpénszintázokat Pfam PF01397 és/vagy PF03936 domének jelenléte alapján is azonosították [139], a Pfam-A.hmm adatbázis ellen hmmscan (HMMER 3.3.2) segítségével [140,141].
A terpén-bioszintézis két útvonalon halad: a kloroplasztikus metil-D-eritritol-foszfát útvonalon, amely monoterpén- és kannabinoid-bioszintézis prekurzorait termeli, valamint a citoszolikus mevalonát útvonalon, amely szeszkviterpén-bioszintézis prekurzorait állítja elő. Ezen útvonalak fehérjeszekvenciáit [142-144] diamond version 2.1.4 segítségével illesztették minden Cannabis proteomhoz [145].
Szintáz-kazetta és kannabinoid-szintáz génanalízis
A 193 kannabiszgenomban a teljes és részleges hosszúságú kannabinoid-szintázok azonosításához a referencia kannabinoid-szintáz szekvenciákat BLASTN-nel illesztették a genomra. A CBDRx-ből fejlesztett, dúsított LTR-szekvenciát további referenciaként használták a szintázok azonosításának segítésére. Az LTR08 a CBDRx-genomból származó, szintáz-kazettákhoz kapcsolódó LTR-szekvencia.
Egy Python-szkript a kannabinoid-szintáz BLAST-eredményeket és LTR08 BLAST-eredményeket táblázatos formában fogadta. Az 500 bp-nál rövidebb szintáz-találatokat kiszűrték. Az 1250-nél kisebb bitscore-ú LTR08-találatokat kiszűrték. A 10-nél kevesebb mismatchet és nulla gapet tartalmazó szintáz- és LTR08-találatokat „Full” szekvenciáknak, az összes többit „Partial” szekvenciának jelölték. Az azonos kezdőpozícióval rendelkező találatokat egyetlen szekvenciára szűrték, majd szintázcímkét kaptak. A szűrt és címkézett szintázokat pályára rajzolták a kannabinoid-szintázok orientációjának vizualizálására; a vizualizációhoz minimum négy szintáz-találat kellett.
A kannabinoid-szintáz génanalízisben először ORFinderrel eltávolították a pszeudogéneket a potenciális gének kezdeti listájából. Ezután usearch11.0.667 segítségével klaszterezték a szintáz kódoló szekvenciákat 0,997 azonossági küszöbbel [146]. A TranslatorX fehérjevezérelt többszörös szekvenciaillesztéseket készített [147]. A szintázok evolúciós történetét a maximum-likelihood módszerrel és General Time Reversible modellel becsülték MEGA11-ben [148].
K-mer kereszteződési elemzés és varin SNP asszociáció
A PanKmer horgonyzási funkcióját ismert kannabisz genotípus-triókban bekövetkező kereszteződési események lokalizálására használták. Tizenegy trió FB191-et, hat trió SSV-t tartalmazott varin-donor szülőként. Az FB191 szülei HO40 és FB30, az SSV szülei HO40 és SSLR voltak; mindkét esetben a HO40 volt a varin donor. Minden trióban az F1 genom haplotípus-felbontott volt, egy haplotípust a varin-donor, egyet a nem varin-donor szülőtől tartalmazva.
Minden esetben PanKmer horgonyzással azonosították a „varin haplotípust”. FB191 trióknál 31-mer indexet készítettek az FB191 genomból alapértelmezett pankmer index paraméterekkel, majd Python-szkripttel, PanKmer API-függvényekkel horgonyozták az indexet minden haplotípusban. A varin haplotípust magasabb 31-mer konzerváció alapján választották ki. Ugyanezt az eljárást alkalmazták SSV triókra is. A lehetséges varin allélokat HO40-ből a keresztezés varin haplotípusáig követték, és a tiszta „haplotípusváltást” jelző k-mer konzervációs értékeknél kereszteződési eseményeket következtettek.
A varin arányadatok kezdetben multimodálisnak tűntek, ezért R-ben a BestNormalize csomag az ordered quantile (ORQ) módszert választotta transzformációra. Ezután R-ben a GAPIT csomag BLINK modelljét használták PCA.total = 6 beállítással az F2 populáció SNP-jei és a transzformált varin arányadatok közötti asszociációk tesztelésére [149]. A PCA.total paramétert QQ-ábrák vizuális értékelése alapján választották ki.
A négy FDR-korrigált szignifikáns SNP körüli régiókban manuálisan értékelték a gén- és TE-modelleket, a k-mer alapú kereszteződési eredményekkel együtt. A négy szignifikáns SNP közül a két legnagyobb magyarázott fenotípusos varianciával társult génekre koncentráltak. A BKR, ALT3 és ALT4 Orthofinder-csoportjait kivonták, az ALT3 és ALT4 ortocsoportokat egyetlen ALT génszámhalmazba vonták össze. A BKR és ALT fehérjeszekvenciák filogéniáit MEGA-ban neighbour-joining módszerrel, 100 bootstrap ismétléssel készítették [148].
Ivari kromoszóma SDR-PAR határ azonosítása és összehasonlítása
Az Y-alapú k-mereket (Y-merek) BWA (v0.7.17) mem segítségével térképezték X/Y haplotípusokra, tökéletes illesztéseket követelve és legfeljebb tízszeres multileképezést engedve. A feltételezett SDR-PAR határok meghatározásához olyan konzervált ortológokat vontak ki, amelyek csökkent Y-mer leképezési sűrűségű régiókban helyezkedtek el, későbbi génfa-elemzéshez. Az ortológokat OrthoFinder (v2.5.4) segítségével definiálták többszörös szekvenciaillesztés opcióval.
Az OrthoFinder minden rendelkezésre álló hím (XY) összeállítás fehérjéivel futott, beleértve a tanulmány hím és több női kontigszintű összeállítását, valamint más vizsgálatok további haplotípus-felbontású összeállításait. Tíz konzervált ortológra, amelyek a feltételezett SDR-PAR határokat fogták át, génfákat becsültek annak megállapítására, hogy az egyes ortológok melyik összeállításban SDR- vagy PAR-kapcsoltak. Ha az Y gametológ szorosan kapcsolódik az SDR-hez, akkor erős támogatás várható külön X- vagy Y-kapcsolt ortológokat tartalmazó kládokra [151].
Mind a tíz konzervált ortológra vagy gametológra blastn (BLAST+ v2.14.1) és bedtools (v2.31.0) getfasta segítségével megkeresték és kivonták a teljes hosszúságú gének nukleotidszekvenciáit intronokkal együtt; a génmátrixokat MAFFT (v7.505) segítségével illesztették –localpair –maxiterate 1000 opciókkal; majd maximum-likelihood fákat becsültek IQ-TREE (v1.6.12) segítségével -MFP -bb 1000 opciókkal.
Az X-Y gametológ fák elemzése után az első feltételezett Y-specifikus, SDR-kapcsolt gén koordinátáit használták az SDR-határok definiálására, majd a kezdő koordinátákat 10 bp-pal kiegészítették. Az X-specifikus régiók kezdetét az X-gametológ koordináták alapján határozták meg az első Y-specifikus génhez viszonyítva. Az SDR-PAR határt az SDR-szegélyrégiókból származó XY gametológ génfák segítségével definiálták, amelyeket hím-specifikus k-merek haplotípusokra térképezésével azonosítottak.
A génfa-elemzés két fő Y-haplotípuscsoportot tárt fel, eltérő SDR-határokkal (Ya és Yb). A „felhőhatár” a kannabiszon belüli SDR-PAR határváltozatosságot jelenti XY gametológ kapcsolatok alapján. A Ya gyakoribb volt az adatkészletben (n = 6), és körülbelül 132 kb-val kiterjesztett SDR-t mutat, amely átfogja a felhőhatárt; e régió a ritkább Yb haplotípusban (n = 2) PAR-kapcsolt marad. A főszövegben közölt Ya haplotípus BCMb (elvadult), GRMa (HC kender), AH3Mb (MJ) és Carmagnola mintában volt megtalálható; a Carmagnola észak-olasz rostkender tájfajta. A Yb haplotípus Kompolti (magyar rostkender-kultivár, amelyet az 1950-es években kiváló rosttulajdonságokra szelektáltak egy régebbi olasz változatból) és GVA-H-21-1003-002 mintában fordult elő.
Jelentési összefoglaló
A kutatási tervről további információ a cikkhez kapcsolt Nature Portfolio Reporting Summary dokumentumban érhető el.
Adatelérhetőség
A kannabisz-pángenom NCBI BioProject azonosítója PRJNA1140642. A pángenom szekvenálási adatai az NCBI Sequence Read Archive (SRA) PRJNA904266 BioProject hozzáférése alatt találhatók. Az EH23a és EH23b BioProject azonosítói PRJNA1111955 és PRJNA1111956. Mind a 193 összeállítás genom- és annotációs fájljai, az orthobrowser és Genome JBrowse példányok, valamint a gráf-pángenomok bemeneti és kimeneti fájljai a https://resources.michael.salk.edu oldalon érhetők el. Az R-gének, terpénszintázok, kannabinoid-szintázok és további genomvizualizációk annotációi a Figshare projektben és DOI-hivatkozások alatt érhetők el; a konkrét genomadatkészletekre mutató linkek az 1. kiegészítő táblázatban találhatók. A forrásadatokat a tanulmány mellékeli.
NCBI BioProject PRJNA1140642: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1140642
SRA BioProject PRJNA904266: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA904266
Erőforrás-portál: https://resources.michael.salk.edu
Figshare Cannabis Pangenome projekt: https://figshare.com/projects/Cannabis_Pangenome/205555
Figshare DOI: https://doi.org/10.25452/figshare.plus.c.7248427.v1
Pangenome metadata and statistics: https://doi.org/10.6084/m9.figshare.25869319.v1
Kódelérhetőség
A szkriptek és elemzési pipeline-ok a GitHubon érhetők el.
CannabisPangenomeShared: https://github.com/anthony-aylward/CannabisPangenomeShared
CannabisPangenomeAnalyses: https://github.com/padgittl/CannabisPangenomeAnalyses
Kiterjesztett adatok ábrái
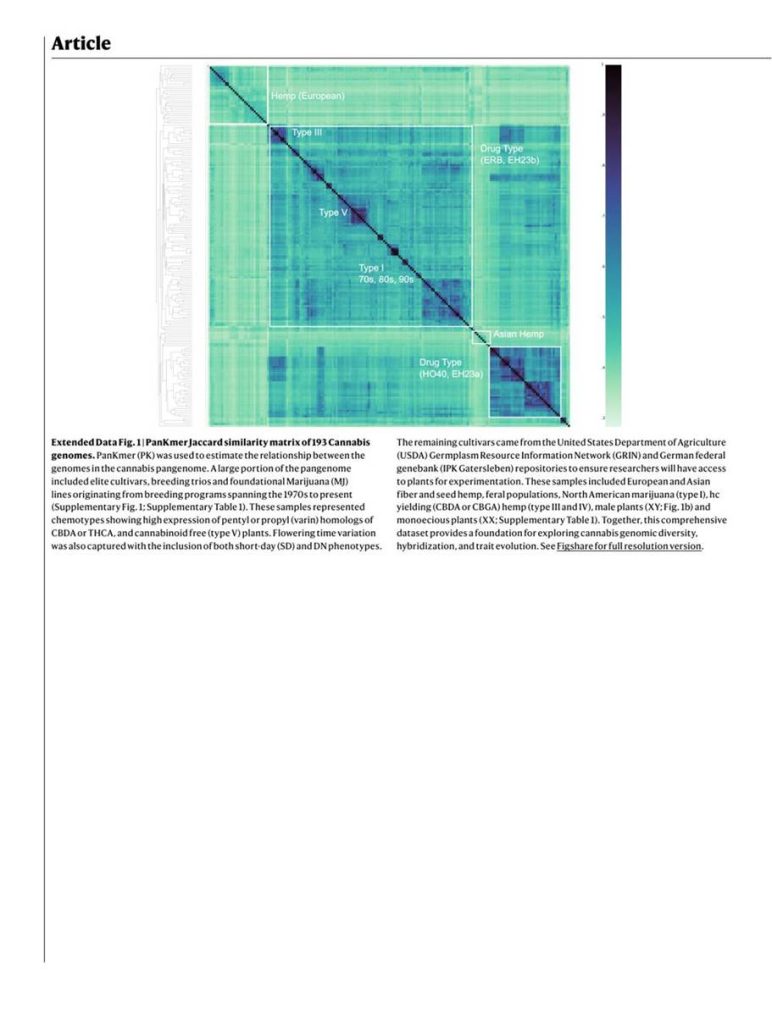
Kiterjesztett adatok 1. ábra | A 193 Cannabis genom PanKmer Jaccard-hasonlósági mátrixa. A PanKmer (PK) segítségével becsülték a kannabisz-pángenomban szereplő genomok közötti kapcsolatot. A pángenom nagy része elit kultivárokat, nemesítési triókat és az 1970-es évektől napjainkig terjedő nemesítési programokból származó alapító marihuána- (MJ) vonalakat tartalmazott. Ezek a minták olyan kemotípusokat képviseltek, amelyek a CBDA vagy THCA pentil- vagy propil- (varin-) homológjainak magas expresszióját mutatták, továbbá kannabinoidmentes (V. típusú) növényeket is tartalmaztak. A virágzási idő változatosságát rövidnappalos (SD) és nappalhossz-semleges (DN) fenotípusok bevonásával is lefedték. A fennmaradó kultivárok az USDA Germplasm Resource Information Network (GRIN) és a német szövetségi génbank (IPK Gatersleben) gyűjteményeiből származtak, hogy a kutatók hozzáférhessenek kísérleti növényanyagokhoz. A minták európai és ázsiai rost- és magkender, elvadult populációk, észak-amerikai marihuána (I. típus), nagy kannabinoidhozamú (CBDA vagy CBGA) kender (III. és IV. típus), hím növények (XY) és egylaki növények (XX) anyagát foglalták magukban. Együttesen ez az átfogó adatkészlet alapot ad a kannabisz genomi diverzitásának, hibridizációjának és tulajdonságevolúciójának vizsgálatához. A teljes felbontású változat a Figshare-en érhető el.

Kiterjesztett adatok 2. ábra | Az EH23 horgonygenom szekvenálási stratégiája és az ebből származó populációk. A) Az F1 hibrid EH23 (ERBxHO40#23) a III. típusú, magas CBDA-tartalmú, nappalhossz-semleges Early Resin Berry (ERB) és az I. típusú, magas THC-tartalmú, nappalérzékeny HO40 keresztezésével jött létre. Az ERB-t és a HO40-et PacBio CLR-rel, míg az EH23-at PacBio HiFi (CCS) technológiával szekvenálták, és nagy áteresztőképességű kromatin-konformáció-befogással (Hi-C) állványozták. Az F2 térképezési populációt (288 egyed) Illumina rövid olvasatokkal szekvenálták. Az OCBD-ből származó fennmaradó pángenom-mintákat a kiegészítő táblázat és a pedigréábra foglalja össze. B) A kannabisz-pángenom 193 genomjának szerveződési sémája. Két módszert használtak haplotípus-felbontású, kromoszómaléptékű genomok előállítására. Az első, egyszerűsített módszer a Hi-C adatokat a fázisoláshoz és az állványozáshoz is felhasználta, és 12 mintából 24 haploid genomot eredményezett (Hifiasm_HiC). Ezek szolgáltak állványozási referenciaként 21 mintából származó 42 genomhoz (Hifiasm_Trio_RagTag), ami triófázisolt haploid összeállításokat eredményezett. Ez a 78 genom együtt a transzponálható elemekre és strukturális variációra vonatkozó pángenomelemzések alapja. Emellett 20 haplotípus-felbontású kontigszintű összeállítást, valamint 83 kontigszintű összeállítást készítettek régebbi PacBio continuous long read (CLR; 23 összeállítás) és circular consensus sequencing (CCS; 60 összeállítás) adatok felhasználásával. C) A tanulmány különböző elemzéseiben használt genomok diagramja. Valamennyi összeállításhoz génmodell-annotációt készítettek ab initio eszközökkel és RNS-expressziós adatokkal, a TE-ket pedig RepeatModeler könyvtár segítségével hívták.
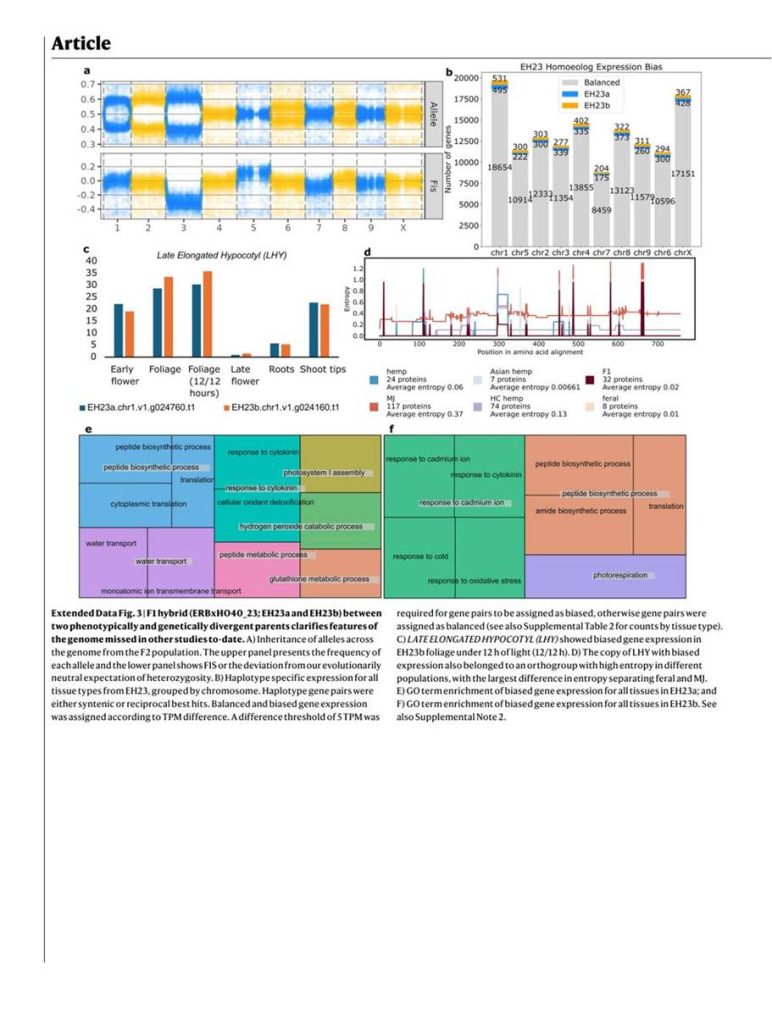
Kiterjesztett adatok 3. ábra | A két fenotípusosan és genetikailag eltérő szülő közötti F1 hibrid (ERBxHO40_23; EH23a és EH23b) olyan genomi jellemzőket tisztáz, amelyeket a korábbi vizsgálatok eddig nem mutattak ki. A) Allélöröklődés a genomban az F2 populáció alapján. A felső panel az egyes allélek gyakoriságát, az alsó panel az FIS értéket, illetve a heterozigozitás evolúciósan semleges várakozásától való eltérést mutatja. B) Haplotípus-specifikus expresszió az EH23 valamennyi szövettípusában, kromoszómánként csoportosítva. A haplotípus génpárok szinténikusak vagy reciprok legjobb találatok voltak. A kiegyensúlyozott és torzított génexpressziót a TPM-különbség alapján rendelték hozzá. A torzított besoroláshoz 5 TPM különbségi küszöböt írtak elő; ellenkező esetben a génpárok kiegyensúlyozottként lettek besorolva. C) A LATE ELONGATED HYPOCOTYL (LHY) torzított génexpressziót mutatott EH23b lombozatban 12 órás fény alatt. D) A torzított expressziót mutató LHY-kópia olyan ortocsoporthoz tartozott, amelynek entrópiája a különböző populációkban magas volt; a legnagyobb entrópiakülönbség az elvadult és az MJ populációkat választotta el. E) A torzított génexpresszió GO-kifejezés-gazdagodása az EH23a valamennyi szövetében; F) a torzított génexpresszió GO-kifejezés-gazdagodása az EH23b valamennyi szövetében.
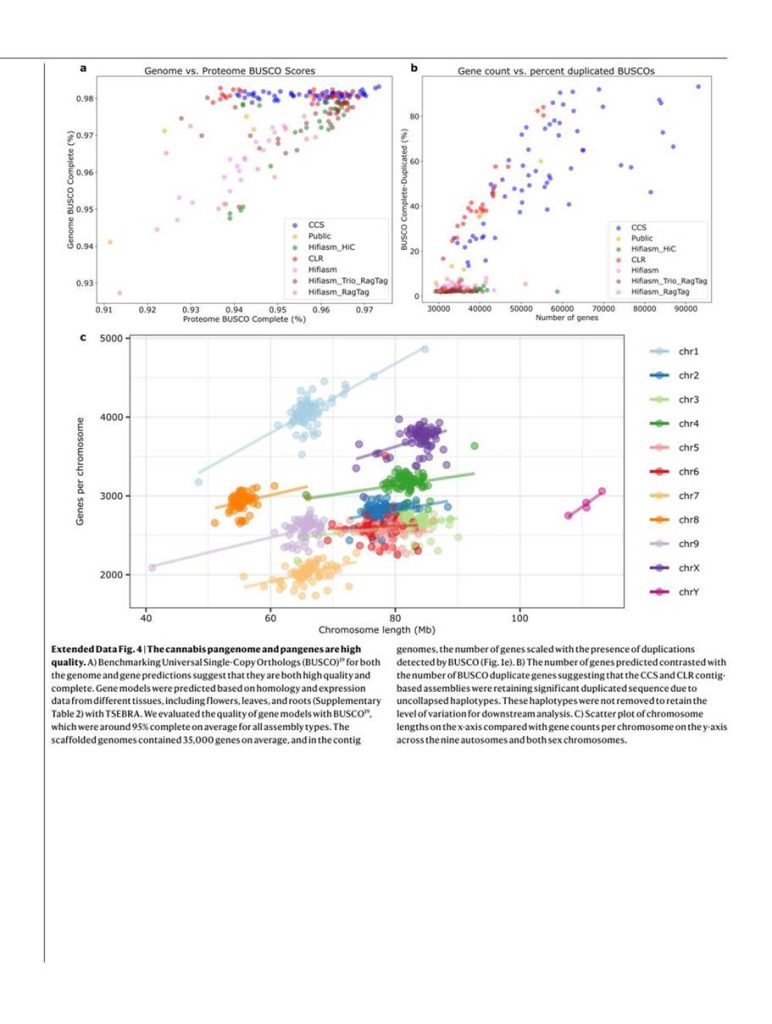
Kiterjesztett adatok 4. ábra | A kannabisz-pángenom és a pángének jó minőségűek. A) A Benchmarking Universal Single-Copy Orthologs (BUSCO) a genomokra és a génelőrejelzésekre egyaránt azt jelzi, hogy azok jó minőségűek és teljesek. A génmodelleket homológia és különböző szövetekből – virágokból, levelekből és gyökerekből – származó expressziós adatok alapján, TSEBRA-val jelezték előre. A génmodellek minőségét BUSCO-val értékelték; átlagosan körülbelül 95%-os teljességet mutattak minden összeállítástípusban. Az állványozott genomok átlagosan 35 000 gént tartalmaztak, a kontiggenomokban pedig a génszám a BUSCO által észlelt duplikációk jelenlétével skálázódott. B) A prediktált gének számának és a BUSCO-duplikált gének számának összevetése arra utal, hogy a CCS- és CLR-kontigalapú összeállítások jelentős duplikált szekvenciát őriztek meg az össze nem omlasztott haplotípusok miatt. Ezeket a haplotípusokat nem távolították el, hogy a későbbi elemzésekhez megőrizzék a variáció szintjét. C) Szórásdiagram: az x tengelyen a kromoszómahosszak, az y tengelyen a kromoszómánkénti génszámok láthatók a kilenc autoszóma és mindkét ivari kromoszóma esetében.

Kiterjesztett adatok 5. ábra | A kannabisz centromer- és telomeranalízise magasabb rendű ismétlésszerkezetet mutat. A-B) Az AceHigh3 (AH3M) kilenc autoszómapárjának és egy ivari kromoszómapárjának (X és Y) kromoszómális jellemzői. Az egymillió bázispáros téglalap-ablakok minden haplotípupárból kifelé nyúlnak, a CpG-motívum hiányával arányos szélességgel. Minden téglalap-ablak génsűrűség szerint színezett; a meleg színek nagy, a hideg színek kis génsűrűséget jeleznek. A haplotípuspárokat poligonok kapcsolják össze, amelyek a szerkezeti elrendezést jelzik: szürke a szinténikus régiók, narancssárga az inverziók esetében. Az egyes haplotípusokon elhelyezett téglalapok kiválasztott lókuszokat jelölnek, beleértve a 45S (26S, 5,8S, 18S) rDNS-tömböket, az 5S RNS-tömböket, a 237 bp-os centromerismétlést, a 370 bp-os CS-1 szubtelomerismétlést és a kannabinoid-szintázokat. Mind a 78 haplotípus-felbontású, kromoszómaléptékű genom kromoszómaábrái hasonló trendeket mutatnak. C) Az AH3M genomban – a pángenom példájaként – Tandem Repeat Finderrel azonosított centromertömbök. Két nagy kópiaszámú tömböt azonosítottak 237 és 370 bp-os bázisismétléssel és ezek magasabb rendű ismétléseivel (HOR). A 237 bp-os tömb ritkán fordul elő a genomban, bár rendszerint a magas „CpG” helyek közelében. A 370 bp-os ismétlés azonos a CS-1 szubtelomerismétléssel, és a kromoszómák végein található. D) A genomok egy részhalmazát Oxford Nanopore Technologies platformon szekvenálták a kannabiszgenomok telomerhosszának becslésére. Az N50 ONT olvasathossz a TeloNum szoftverrel azonosított maximális telomerismétlés függvényében látható.
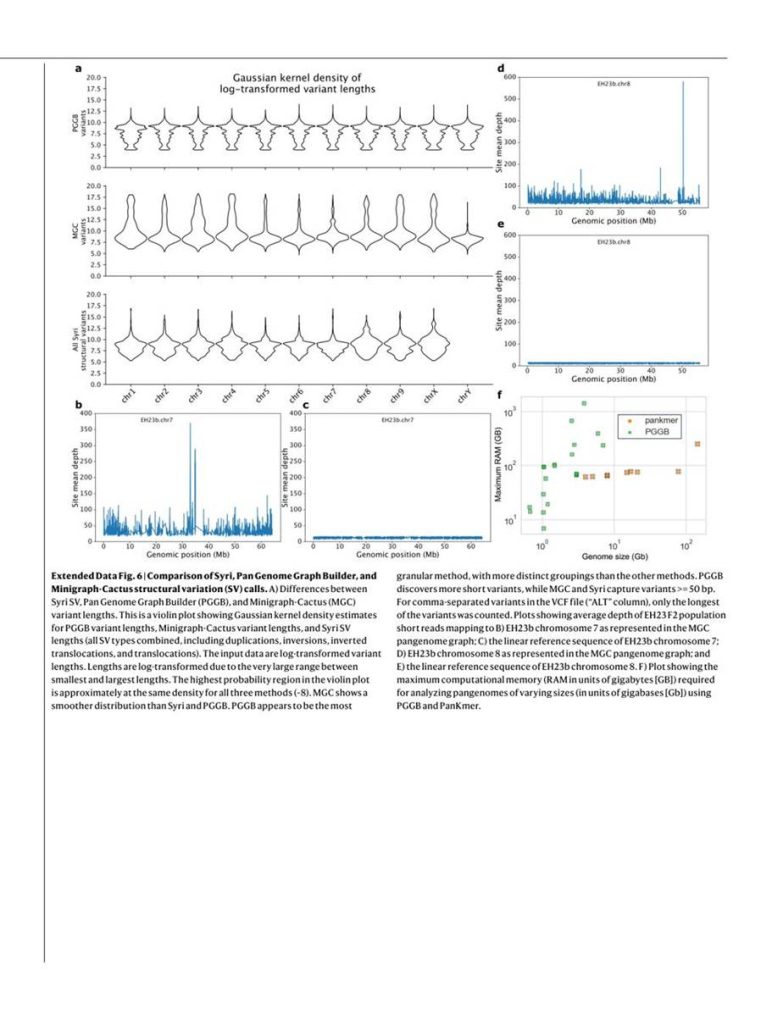
Kiterjesztett adatok 6. ábra | A Syri, a Pan Genome Graph Builder és a Minigraph-Cactus strukturális variáns (SV) hívásainak összehasonlítása. A) A Syri SV-, a Pan Genome Graph Builder (PGGB)- és a Minigraph-Cactus (MGC)-variánshosszok közötti különbségek. A hegedűdiagram Gauss-féle kernel-sűrűségbecslést mutat a PGGB-, MGC- és Syri-SV variánshosszokra; minden SV-típus szerepel benne, beleértve a duplikációkat, inverziókat, invertált transzlokációkat és transzlokációkat. A bemeneti adatok log-transzformált variánshosszok, mivel a legkisebb és legnagyobb hosszok között igen nagy a tartomány. A hegedűdiagram legnagyobb valószínűségű régiója mindhárom módszernél közel azonos sűrűségnél található. Az MGC eloszlása simább, mint a Syrié és a PGGB-é. A PGGB tűnik a leggranulárisabb módszernek, több elkülönült csoporttal, mint a többi módszer. A PGGB több rövid variánst fedez fel, míg az MGC és a Syri az 50 bp-nál nagyobb vagy azzal egyenlő variánsokat ragadja meg. Vesszővel elválasztott VCF-variánsok esetében csak a leghosszabb variánst számolták. Az EH23 F2 populáció rövid olvasatainak átlagos mélységét mutató ábrák: B) EH23b 7. kromoszóma az MGC pángenomgráfban; C) az EH23b 7. kromoszóma lineáris referenciaszekvenciája; D) EH23b 8. kromoszóma az MGC pángenomgráfban; E) az EH23b 8. kromoszóma lineáris referenciaszekvenciája. F) A különböző méretű pángenomok elemzéséhez PGGB-vel és PanKmerrel szükséges maximális számítási memória (RAM, GB-ban), a pángenom méretének (Gb) függvényében.

Kiterjesztett adatok 7. ábra | Terpénszintáz gének a kannabisz-pángenomban. A) Hegedűdiagram a terpénszintáz-kópiaszámról a kannabisz-pángenomban. Az 5. és 6. kromoszóma kópiaszám-forrópont a kannabisz-pángenomban. B) Az EH23a.chr6.v1.g321150.t1 – az EH23a 6. kromoszómáján valamennyi virágmintában legmagasabban expresszált terpénszintáz – Odgi 2D vizualizációja referenciafüggetlen PGGB-pángenomgráfból. C) Az EH23a.chr6.v1.g321150.t1 pángenom-variációs gráf-vizualizációja, amely a génszekvencia mentén elszórt variációs régiókat mutat. D) A fehérje többszörös szekvenciaillesztésének entrópiaértékei: az illesztés elején alacsony, a vége felé magas variáció látható.
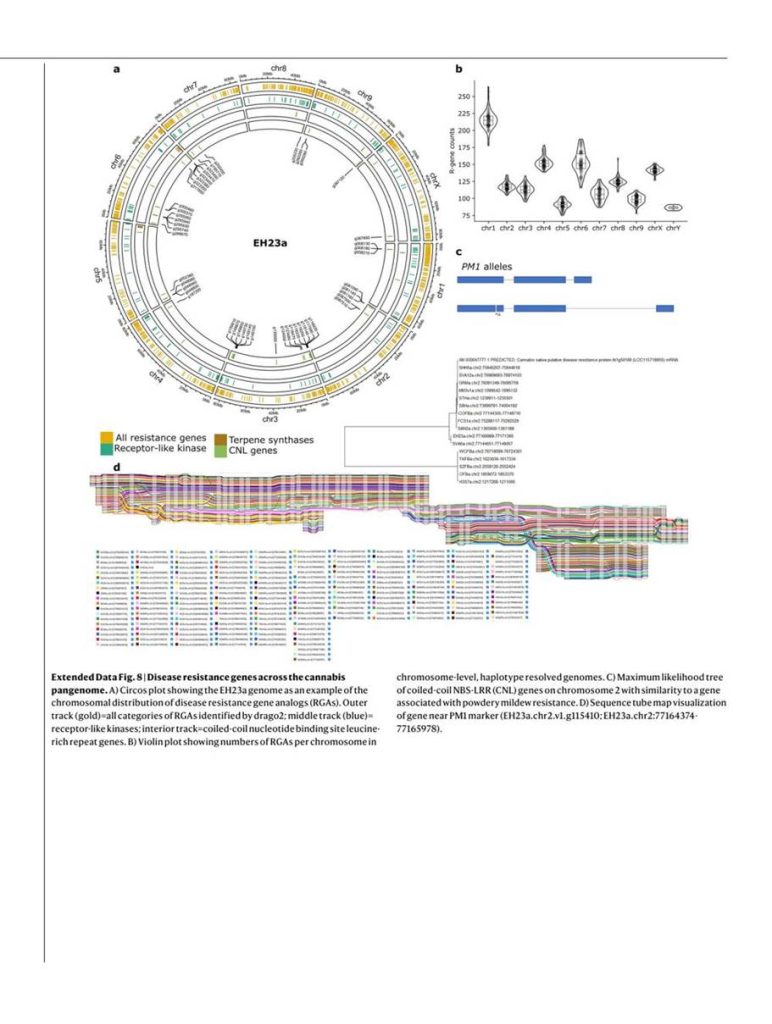
Kiterjesztett adatok 8. ábra | Betegségrezisztencia gének a kannabisz-pángenomban. A) Circos-ábra az EH23a genom példáján a betegségrezisztencia-génanalógok (RGA-k) kromoszómális eloszlásáról. Külső sáv (arany): a drago2 által azonosított RGA-k minden kategóriája; középső sáv (kék): receptor-like kinase-ok; belső sáv: coiled-coil nucleotide-binding site leucine-rich repeat gének. B) Hegedűdiagram a kromoszómánkénti RGA-számokról kromoszómaszintű, haplotípus-felbontású genomokban. C) Maximum-likelihood fa a 2. kromoszómán található coiled-coil NBS-LRR (CNL) génekről, amelyek hasonlóságot mutatnak egy lisztharmat-rezisztenciához kapcsolt génnel. D) A PM1 marker közelében lévő gén szekvencia-csőtérkép-vizualizációja.
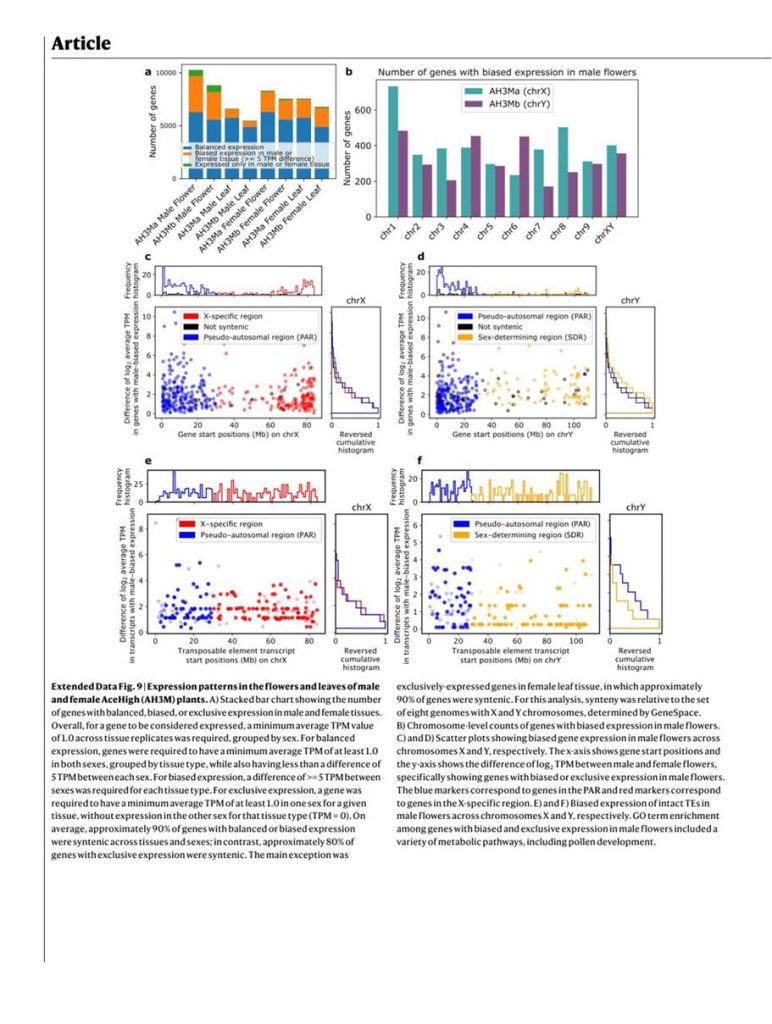
Kiterjesztett adatok 9. ábra | Hím és nőivarú AceHigh (AH3M) növények virágaiban és leveleiben megfigyelt expressziós mintázatok. A) Halmozott oszlopdiagram a hím és női szövetekben kiegyensúlyozott, torzított vagy kizárólagos expressziót mutató gének számáról. Általánosságban ahhoz, hogy egy gén expresszáltnak minősüljön, legalább 1,0 átlagos TPM értékre volt szükség a szöveti ismétlések között, ivar szerint csoportosítva. Kiegyensúlyozott expresszió esetén a géneknek legalább 1,0 átlagos TPM-et kellett mutatniuk mindkét ivarban, szövettípus szerint csoportosítva, és a két ivar között 5 TPM-nél kisebb különbséget kellett mutatniuk. Torzított expresszió esetén szövettípusonként legalább 5 TPM különbségre volt szükség az ivarok között. Kizárólagos expresszió esetén a génnek egy adott szövetben legalább 1,0 átlagos TPM-et kellett mutatnia az egyik ivarban, miközben a másik ivarban ugyanabban a szövettípusban nem volt expresszió (TPM = 0). Átlagosan a kiegyensúlyozott vagy torzított expressziót mutató gének körülbelül 90%-a volt szinténikus a szövetek és ivarok között; ezzel szemben a kizárólagos expressziót mutató gének körülbelül 80%-a volt szinténikus. A fő kivételt a nőivarú levélszövetben kizárólagosan expresszált gének jelentették, amelyeknek körülbelül 90%-a volt szinténikus. Ebben az elemzésben a szinténia a GeneSpace által meghatározott, X és Y kromoszómát tartalmazó nyolc genom halmazához viszonyított. B) Hímvirágokban torzított expressziót mutató gének kromoszómaszintű száma. C) és D) Szórásdiagramok a hímvirágokban torzított génexpresszióról az X, illetve Y kromoszómán. Az x tengely a génkezdő pozíciókat, az y tengely a hím és női virágok közötti log2 TPM-különbséget mutatja, kifejezetten a hímvirágokban torzított vagy kizárólagos expressziót mutató génekre. A kék markerek a PAR-ban, a vörös markerek az X-specifikus régióban található géneknek felelnek meg. E) és F) Intakt TE-k torzított expressziója hímvirágokban az X, illetve Y kromoszómán. A hímvirágokban torzított és kizárólagos expressziót mutató gének között gazdagodó GO-kifejezések különféle metabolikus útvonalakat, köztük pollenfejlődést tartalmaztak.

Kiterjesztett adatok 10. ábra | A kannabisz-pángenom a strukturális variációk (SV-k) széles tartományát tárja fel, amely némely értékében fajok közötti összehasonlításokban közölt értékekkel is összemérhető. A) Három SV-típus eloszlása a kannabisz-pángenom 78 állványozott összeállításában. Minden mintaösszeállítást az EH23a haplotípus-összeállításhoz illesztettek SV-híváshoz. B) Az inverzióhosszak többcsúcsú eloszlása az összes minta minden inverziójára. C) Az egyes összeállításokban található inverziók teljes hosszának eloszlása a teljes genom hosszának százalékaként. D) Az inverzióhosszak eloszlása az összes minta minden inverziójára. E) Kódoló szekvenciák (CDS-ek) és intakt transzponálható elemek (TE-k) eloszlása valamennyi inverzióban és szinténikus régióban, mintánként. Az inverziók a szinténikus régiókhoz képest szignifikánsan kevesebb CDS-t tartalmaznak, míg a TE-k átlagosan közel azonos szinten vannak jelen az inverziókban és a szinténikus régiókban. F) Az inverziós töréspontpárok – amelyeket minden 10 kb-nál nagyobb inverzió kezdő- és végpontja köré központosított 8 kb-os ablakokként definiáltak – az esetek mintegy 50%-ában repetitív elemeket tartalmaznak. G) Az inverziós töréspontok önmagukhoz illesztve nagyobb arányban mutatnak szegmentális duplikációkat, de kisebb arányban invertált ismétléseket, mint a kezdő-végpont páros illesztések. F) Példa egy európai kender minta haplotípusának (KC Dora) illesztésére és SV-ire. A két megabázis-léptékű inverzió a 4. kromoszóma olyan régiójában található, amely korábbi, elvadult amerikai kender és marihuána-populációkat összehasonlító munkában emelkedett Fst-értékeket mutatott [157].
Köszönetnyilvánítás
A szerzők köszönetet mondanak a Michael-laboratórium tagjainak a munka megvitatásáért; továbbá T. Gordonnak és Z. Stansellnek a GRIN-gyűjteményből származó vonalak levélanyagának elküldéséért.
Tanulmány-információk
Eredeti közlemény
Lynch RC, Padgitt-Cobb LK, Garfinkel AR és munkatársai (2025) Domesticated cannabinoid synthases amid a wild mosaic cannabis pangenome. Nature 643: 1001–1010. DOI: 10.1038/s41586-025-09065-0
Szerzői információk
Szerzők: Ryan C. Lynch, Lillian K. Padgitt-Cobb, Andrea R. Garfinkel, Brian J. Knaus, Nolan T. Hartwick, Nicholas Allsing, Anthony Aylward, Philip C. Bentz, Sarah B. Carey, Allen Mamerto, Justine K. Kitony, Kelly Colt, Emily R. Murray, Tiffany Duong, Heidi I. Chen, Aaron Trippe, Alex Harkess, Seth Crawford, Kelly Vining és Todd P. Michael
Egyenlő szerzői hozzájárulás: Ryan C. Lynch és Lillian K. Padgitt-Cobb egyenlő mértékben járultak hozzá a munkához.
Levelezés és anyagkérések: Ryan C. Lynch, Lillian K. Padgitt-Cobb vagy Todd P. Michael.
Kapcsolat: rlynch@colorado.edu; lilliankpc@gmail.com; toddpmichael@gmail.com
Előzménydátumok
Érkezett: 2024. május 21.; elfogadva: 2025. április 24.; online megjelent: 2025. május 28.; nyomtatott lapszám: Nature 643, 1001–1010, 2025. július 24.
Szerzői hozzájárulások
T.P.M., R.C.L., S.C., A.R.G., K.V. és L.K.P.-C. tervezték és szervezték a kutatási erőfeszítéseket. R.C.L., L.K.P.-C., T.P.M., B.J.K., N.T.H., N.A., A.A., A.M., J.K.K., H.I.C., A.R.G., A.T., P.C.B., S.B.C. és A.H. elemezték a pángenomadatokat. R.C.L., L.K.P.-C., A.R.G., T.P.M., K.C., E.R.M., T.D. és S.C. végeztek üvegházi, szántóföldi és laboratóriumi kísérleteket. R.C.L., L.K.P.-C., T.P.M., B.J.K. és K.V. írták és szerkesztették a kéziratot. R.C.L., L.K.P.-C. és T.P.M. javították a kéziratot. Minden szerző elolvasta és jóváhagyta a kéziratot.
Versengő érdekek
S.C. az Oregon CBD társalapítója volt. A.R.G. és A.T. az Oregon CBD alkalmazottai voltak. R.C.L. érdekelt a Saint Vrain Research LLC-ben, amely kenderalapú termékeket gyárt. T.P.M. a CQuesta szénmegkötő vállalat alapítója. A.H. a Veil Genomics genotipizáló vállalat társalapítója. A többi szerző nem nyilatkozott versengő érdekről.
Finanszírozási információk
A munkát részben a Tang genomikai alap (T.P.M.), L.K.P.-C. National Science Foundation Plant Genome Postdoctoral Research Fellowship támogatása (NSF-IOS PRFB 2209290), valamint a pángenomeszközök Michael-laboratóriumbeli fejlesztéséhez a Bill and Melinda Gates Foundation (INV-040541) (T.P.M.) támogatta. További támogatást nyújtott a US Department of Agriculture National Institute of Food and Agriculture Postdoctoral Fellowship (USDA NIFA) 2022-67012-38987 (S.B.C.), az USDA NIFA 2023-67013-39620 (A.H.) és a National Science Foundation (NSF) IOS-PGRP CAREER 2239530 (A.H.).
Szakmai bírálat és kiadói megjegyzés
A Nature köszönetet mond Shelby Ellisonnak, Manuel Spannaglnak és a további anonim bírálóknak a kézirat szakmai bírálatához nyújtott hozzájárulásukért; a bírálati jelentések elérhetők. A kiadói megjegyzés szerint a Springer Nature semleges marad a közzétett térképek és intézményi hovatartozások joghatósági állításaival kapcsolatban.
Szerzői jog és licenc
Az eredeti cikk Creative Commons Attribution 4.0 International License (CC BY 4.0) alatt jelent meg, amely lehetővé teszi a használatot, megosztást, adaptációt, terjesztést és reprodukciót bármilyen médiumban vagy formátumban, feltéve, hogy megfelelően feltüntetik az eredeti szerzőket és a forrást, linket adnak a licenchez, és jelzik, ha változtatás történt. A képek vagy harmadik féltől származó anyagok a cikk CC-licencébe tartoznak, kivéve, ha a képaláírás vagy kreditvonal másként jelzi.
Adat-, kód- és kiegészítő anyagok
Jelentési összefoglaló
A kutatási tervről további információ a cikkhez kapcsolt Nature Portfolio Reporting Summary dokumentumban érhető el.
Adatelérhetőség
A kannabisz-pángenom NCBI BioProject azonosítója PRJNA1140642. A pángenom szekvenálási adatai az NCBI Sequence Read Archive (SRA) PRJNA904266 BioProject hozzáférése alatt találhatók. Az EH23a és EH23b BioProject azonosítói PRJNA1111955 és PRJNA1111956. Mind a 193 összeállítás genom- és annotációs fájljai, az orthobrowser és Genome JBrowse példányok, valamint a gráf-pángenomok bemeneti és kimeneti fájljai a Michael-laboratórium erőforrásportálján érhetők el.
NCBI BioProject PRJNA1140642: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1140642
SRA BioProject PRJNA904266: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA904266
Erőforrás-portál: https://resources.michael.salk.edu
Figshare Cannabis Pangenome projekt: https://figshare.com/projects/Cannabis_Pangenome/205555
Figshare DOI: 10.25452/figshare.plus.c.7248427.v1
Pangenome metadata and statistics: 10.6084/m9.figshare.25869319.v1
Kódelérhetőség
A szkriptek és elemzési pipeline-ok a GitHubon érhetők el.
CannabisPangenomeShared: https://github.com/anthony-aylward/CannabisPangenomeShared
CannabisPangenomeAnalyses: https://github.com/padgittl/CannabisPangenomeAnalyses
Kiegészítő információk és forrásadatok
Supplementary Information (PDF): PDF megnyitása
Reporting Summary (PDF): PDF megnyitása
Peer Review file (PDF): PDF megnyitása
Source Data Fig. 1: XLSX letöltése
Source Data Fig. 2: XLSX letöltése
Source Data Fig. 4: XLSX letöltése
Hivatkozások
A hivatkozásjegyzék bibliográfiai adatai az eredeti cikkből változatlanul átvéve szerepelnek; a szövegben szereplő számozott hivatkozások közvetlenül a Nature online hivatkozási horgonyaira mutatnak.
1. Long, T., Wagner, M., Demske, D., Leipe, C. & Tarasov, P. E. Cannabis in Eurasia: origin of human use and Bronze Age trans-continental connections. Veg. Hist. Archaeobot. 26, 245–258 (2017).
2. Ren, G. et al. Large-scale whole-genome resequencing unravels the domestication history of Cannabis sativa. Sci. Adv. 7, eabg2286 (2021).
3. Bai, Y. et al. Archaeobotanical evidence of the use of medicinal cannabis in a secular context unearthed from south China. J. Ethnopharmacol. 275, 114114 (2021).
4. Kovalchuk, I. et al. The genomics of cannabis and its close relatives. Annu. Rev. Plant Biol. 71, 713–739 (2020).
5. Clarke, R. & Merlin, M. Cannabis: Evolution and Ethnobotany (Univ. of California Press, 2016).
6. Stoa, R. Craft Weed: Family Farming and the Future of the Marijuana Industry (MIT Press, 2018).
7. Patton, D. V. A history of United States cannabis law. J. Law Health 34, 1–29 (2020).
8. Bewley-Taylor, D. & Jelsma, M. Regime change: re-visiting the 1961 Single Convention on Narcotic Drugs. Int. J. Drug Policy 23, 72–81 (2012).
9. Hanuš, L. O., Meyer, S. M., Muñoz, E., Taglialatela-Scafati, O. & Appendino, G. Phytocannabinoids: a unified critical inventory. Nat. Prod. Rep. 33, 1357–1392 (2016).
10. Devinsky, O. et al. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N. Engl. J. Med. 376, 2011–2020 (2017).
11. Grassa, C. J. et al. A new Cannabis genome assembly associates elevated cannabidiol (CBD) with hemp introgressed into marijuana. New Phytol. 230, 1665–1679 (2021).
12. McKernan, K. J. et al. Sequence and annotation of 42 cannabis genomes reveals extensive copy number variation in cannabinoid synthesis and pathogen resistance genes. Preprint at bioRxiv https://doi.org/10.1101/2020.01.03.894428 (2020).
13. Gao, S. et al. A high-quality reference genome of wild Cannabis sativa. Hortic. Res. 7, 73 (2020).
14. Braich, S., Baillie, R. C., Spangenberg, G. C. & Cogan, N. O. I. A new and improved genome sequence of Cannabis sativa. GigaByte https://doi.org/10.46471/gigabyte.10 (2020).
15. van Bakel, H. et al. The draft genome and transcriptome of Cannabis sativa. Genome Biol. 12, R102 (2011).
16. Laverty, K. U. et al. A physical and genetic map of Cannabis sativa identifies extensive rearrangements at the THC/CBD acid synthase loci. Genome Res. 29, 146–156 (2019).
17. Barcaccia, G. et al. Potentials and challenges of genomics for breeding cannabis cultivars. Front. Plant Sci. 11, 573299 (2020).
18. McPartland, J. M. & Small, E. A classification of endangered high-THC cannabis (Cannabis sativa subsp. indica) domesticates and their wild relatives. PhytoKeys 144, 81–112 (2020).
19. Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
20. Hickey, G. et al. Pangenome graph construction from genome alignments with Minigraph- Cactus. Nat. Biotechnol. 42, 663–673 (2024).
21. Aylward, A. J., Petrus, S., Mamerto, A., Hartwick, N. T. & Michael, T. P. PanKmer: k-mer- based and reference-free pangenome analysis. Bioinformatics 39, btad621 (2023).
22. Garrison, E. et al. Building pangenome graphs. Nat. Methods 21, 2008–2012 (2024).
23. McPartland, J. M. & Guy, G. W. Models of cannabis taxonomy, cultural bias, and conflicts between scientific and vernacular names. Bot. Rev. 83, 327–381 (2017).
24. Qiao, Q. et al. Evolutionary history and pan-genome dynamics of strawberry (Fragaria spp.). Proc. Natl Acad. Sci. USA 118, e2105431118 (2021).
25. Li, C., Lin, H., Debernardi, J. M., Zhang, C. & Dubcovsky, J. GIGANTEA accelerates wheat heading time through gene interactions converging on FLOWERING LOCUS T1. Plant J. 118, 519–533 (2024).
26. Steed, G., Ramirez, D. C., Hannah, M. A. & Webb, A. A. R. Chronoculture, harnessing the circadian clock to improve crop yield and sustainability. Science 372, eabc9141 (2021).
27. de Meijer, E. Fibre hemp cultivars: a survey oforigin, ancestry,availability and brief agronomic characteristics. J. Int. Hemp Assoc. 2, 66–73 (1995).
28. Westergaard. M. in Advances in Genetics, Vol. 9 (ed. Demerec, M.) 217–281 (Academic Press, 1958).
29. Carey, S. B. et al. The evolution of heteromorphic sex chromosomes in plants. Preprint at bioRxiv https://doi.org/10.1101/2024.12.09.627636 (2024).
30. McPartland, J. M. Cannabis systematics at the levels of family, genus, and species. Cannabis Cannabinoid Res. 3, 203–212 (2018).
31. Prentout, D. et al. Plant genera Cannabis and Humulus share the same pair of well- differentiated sex chromosomes. New Phytol. 231, 1599–1611 (2021).
32. Petit, J., Salentijn, E. M. J., Paulo, M.-J., Denneboom, C. & Trindade, L. M. Genetic architecture of flowering time and sex determination in hemp (Cannabis sativa L.): a genome-wide association study. Front. Plant Sci. 11, 569958 (2020).
33. Charlesworth, D., Charlesworth, B. & Marais, G. Steps in the evolution of heteromorphic sex chromosomes. Heredity 95, 118–128 (2005).
34. Stack, G. M. et al. Comparison of recombination rate, reference bias, and unique pangenomic haplotypes in Cannabis sativa using seven de novo genome assemblies. Int. J. Mol. Sci. 26, 1165 (2025).
35. Lu, C. et al. Phosphorylation of SPT5 by CDKD;2 is required for VIP5 recruitment and normal flowering in Arabidopsis thaliana. Plant Cell 29, 277–291 (2017).
36. Lappin, F. M. et al. A polymorphic pseudoautosomal boundary in the Carica papaya sex chromosomes. Mol. Genet. Genomics 290, 1511–1522 (2015).
37. Grabowska-Joachimiak, A., Śliwińska, E., Piguła, M., Skomra, U. & Joachimiak, A. J. Genome size in Humulus lupulus L. and H. japonicus Siebold and Zucc. (Cannabaceae). Acta Soc. Bot. Pol. 75, 207–214 (2006).
38. Ma, J., Devos, K. M. & Bennetzen, J. L. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 14, 860–869 (2004).
39. Choi, J., Lyons, D. B., Kim, M. Y., Moore, J. D. & Zilberman, D. DNA methylation and histone H1 jointly repress transposable elements and aberrant intragenic transcripts. Mol. Cell 77, 310–323.e7 (2020).
40. Harringmeyer, O. S. & Hoekstra, H. E. Chromosomal inversion polymorphisms shape the genomic landscape of deer mice. Nat. Ecol. Evol. 6, 1965–1979 (2022).
41. Hirabayashi, K. & Owens, G. L. The rate of chromosomal inversion fixation in plant genomes is highly variable. Evolution 77, 1117–1130 (2023).
42. Gabur, I., Chawla, H. S., Snowdon, R. J. & Parkin, I. A. P. Connecting genome structural variation with complex traits in crop plants. Züchter Genet. Breed. Res. 132, 733–750 (2019).
43. Jay, P. et al. Supergene evolution triggered by the introgression of a chromosomal inversion. Curr. Biol. 28, 1839–1845.e3 (2018).
44. Toth, J. A., Stack, G. M., Carlson, C. H. & Smart, L. B. Identification and mapping of major-effect flowering time loci Autoflower1 and Early1 in Cannabis sativa L. Front. Plant Sci. 13, 991680 (2022).
45. Murphy, R. L. et al. Coincident light and clock regulation of pseudoresponse regulator protein 37 (PRR37) controls photoperiodic flowering in sorghum. Proc. Natl Acad. Sci. USA 108, 16469–16474 (2011).
46. Li, M.-W., Liu, W., Lam, H.-M. & Gendron, J. M. Characterization of two growth period QTLs reveals modification of PRR3 genes during soybean domestication. Plant Cell Physiol. 60, 407–420 (2019).
47. Whiting, J. R. et al. The genetic architecture of repeated local adaptation to climate in distantly related plants. Nat. Ecol. Evol. 8, 1933–1947 (2024).
48. Todesco, M. et al. Massive haplotypes underlie ecotypic differentiation in sunflowers. Nature 584, 602–607 (2020).
49. Andre, C. M. et al. Unique bibenzyl cannabinoids in the liverwort Radula marginata: parallels with Cannabis chemistry. New Phytol. https://doi.org/10.1111/nph.20349 (2024).
50. van Velzen, R. & Schranz, M. E. Origin and evolution of the cannabinoid oxidocyclase gene family. Genome Biol. Evol. 13, evab130 (2021).
51. Smith, C. J., Vergara, D., Keegan, B. & Jikomes, N. The phytochemical diversity of commercial Cannabis in the United States. PLoS ONE 17, e0267498 (2022).
52. de Meijer, E. P. M. & Hammond, K. M. The inheritance of chemical phenotype in Cannabis sativa L. (V): regulation of the propyl-/pentyl cannabinoid ratio, completion of a genetic model. Euphytica 210, 291–307 (2016).
53. Vigli, D. et al. Chronic treatment with the phytocannabinoid Cannabidivarin (CBDV) rescues behavioural alterations and brain atrophy in a mouse model of Rett syndrome. Neuropharmacology 140, 121–129 (2018).
54. Welling, M. T. et al. An extreme-phenotype genome‐wide association study identifies candidate cannabinoid pathway genes in Cannabis. Sci. Rep. 10, 18643 (2020).
55. Pulsifer, I. P. et al. Acyl-lipid thioesterase1-4 from Arabidopsis thaliana form a novel family of fatty acyl-acyl carrier protein thioesterases with divergent expression patterns and substrate specificities. Plant Mol. Biol. 84, 549–563 (2014).
56. Kalinger, R. S., Pulsifer, I. P., Hepworth, S. R. & Rowland, O. Fatty acyl synthetases and thioesterases in plant lipid metabolism: diverse functions and biotechnological applications. Lipids 55, 435–455 (2020).
57. Turner, C. E. et al. Constituents of Cannabis sativa L. IV. Stability of cannabinoids in stored plant material. J. Pharm. Sci. 62, 1601–1605 (1973).
58. Welling, M. T., Liu, L., Shapter, T., Raymond, C. A. & King, G. J. Characterisation of cannabinoid composition in a diverse Cannabis sativa L. germplasm collection. Euphytica 208, 463–475 (2016).
59. Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
60. Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
61. Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
62. Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
63. Krueger, F. et al. FelixKrueger/TrimGalore: v0.6.10. Zenodo https://doi.org/10.5281/ zenodo.7598955 (2023).
64. Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. Preprint at https://doi.org/10.48550/arXiv.1207.3907 (2012).
65. Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
66. Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
67. Garfinkel, A. R., Otten, M. & Crawford, S. SNP in potentially defunct tetrahydrocannabinolic acid synthase is a marker for cannabigerolic acid dominance in Cannabis sativa L. Genes 12, 228 (2021).
68. Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
69. Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
70. Hunter, Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 9, 90–95 (2007).
71. Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
72. Harris, C. R. et al. Array programming with NumPy. Nature 585, 357–362 (2020).
73. Alexa, A. & Rahnenführer, J. topGO: enrichment analysis for Gene Ontology. https://doi. org/10.18129/B9.bioc.topGO, R package version 2.59.0 (2024).
74. Denyer, T. et al. Streamlined spatial and environmental expression signatures characterize the minimalist duckweed Wolffia australiana. Genome Res. 34, 1106–1120 (2024).
75. Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
76. Schalamun, M. High molecular weight gDNA extraction after Mayjonade et al. optimised for eucalyptus for nanopore sequencing. Protocols.io https://doi.org/10.17504/protocols. io.i6vche6 (2017).
77. Chin, C.-S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054 (2016).
78. Nurk, S. et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30, 1291–1305 (2020).
79. Titus Brown, C. & Irber, L. sourmash: a library for MinHash sketching of DNA. J. Open Source Softw. 1, 27 (2016).
80. Alonge, M. et al. Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome Biol. 23, 258 (2022).
81. Kurtzer, G. M. et al. Hpcng/singularity: Singularity 3.7.1. Zenodo https://doi.org/10.5281/ ZENODO.4435194 (2021).
82. Garrison, E. et al. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 36, 875–879 (2018).
83. Liao, W.-W. et al. A draft human pangenome reference. Nature 617, 312–324 (2023).
84. Di Tommaso, P. et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 35, 316–319 (2017).
85. Heumos, S. et al. Cluster-efficient pangenome graph construction with nf-core/pangenome. Bioinformatics 40, btae609 (2024).
86. Heumos, S. et al. Nf-core/pangenome: Pangenome 1.1.2 – canguro. Zenodo https://doi.org/
10. 5281/ZENODO.10869589 (2024).
87. Sirén, J. et al. Pangenomics enables genotyping of known structural variants in 5202 diverse genomes. Science 374, abg8871 (2021).
88. Hickey, G. et al. Genotyping structural variants in pangenome graphs using the vg toolkit. Genome Biol. 21, 35 (2020).
89. Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl Acad. Sci. USA 117, 9451–9457 (2020).
90. Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
91. Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. 2013−2015; https://www. repeatmasker.org/ (2015).
92. Gabriel, L., Hoff, K. J., Brůna, T., Borodovsky, M. & Stanke, M. TSEBRA: transcript selector for BRAKER. BMC Bioinformatics 22, 566 (2021).
93. Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
94. Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
95. Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG- mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
96. Waterhouse, R. M. et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
97. Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275 (2019).
98. Goel, M., Sun, H., Jiao, W.-B. & Schneeberger, K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 20, 277 (2019).
99. Goel, M. & Schneeberger, K. plotsr: visualizing structural similarities and rearrangements between multiple genomes. Bioinformatics 38, 2922–2926 (2022).
100. Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
101. VanBuren, R. et al. Single-molecule sequencing of the desiccation-tolerant grass Oropetium thomaeum. Nature 527, 508–511 (2015).
102. Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
103. Colt, K. et al. Telomere length in plants estimated with long read sequencing. Preprint at bioRxiv https://doi.org/10.1101/2024.03.27.586973 (2024).
104. Garcia-Cisneros, A. et al. Long telomeres are associated with clonality in wild populations of the fissiparous starfish Coscinasterias tenuispina. Heredity 115, 480 (2015).
105. Melters, D. P. et al. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 14, R10 (2013).
106. Divashuk, M. G., Alexandrov, O. S., Razumova, O. V., Kirov, I. V. & Karlov, G. I, Molecular cytogenetic characterization of the dioecious Cannabis sativa with an XY chromosome sex determination system. PLoS ONE 9, e85118 (2014).
107. Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
108. Knaus, B. J. & Grünwald, N. J. vcfr: a package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 17, 44–53 (2017).
109. Wright, S. The genetical structure of populations. Ann. Eugen. 15, 323–354 (1951).
110. Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 11, e0163962 (2016).
111. Kaur, H., Shannon, L. M. & Samac, D. A. A stepwise guide for pangenome development in crop plants: an alfalfa (Medicago sativa) case study. BMC Genomics 25, 1022 (2024).
112. Koch, M. A., Haubold, B. & Mitchell-Olds, T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol. Biol. Evol. 17, 1483–1498 (2000).
113. Lynch, M. & Conery, J. S. The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155 (2000).
114. Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
115. Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126 (2018).
116. Pereira, V. Insertion bias and purifying selection of retrotransposons in the Arabidopsis thaliana genome. Genome Biol. 5, R79 (2004).
117. VanBuren, R. et al. Extreme haplotype variation in the desiccation-tolerant clubmoss Selaginella lepidophylla. Nat. Commun. 9, 13 (2018).
118. Karakülah, G. & Suner, A. PlanTEnrichment: a tool for enrichment analysis of transposable elements in plants. Genomics 109, 336–340 (2017).
119. Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289–300 (1995).
120. Seabold, S. & Perktold, J. Statsmodels: econometric and statistical modeling with Python. In Proc. 9th Python in Science Conference https://doi.org/10.25080/Majora-92bf1922-011 (SciPy, 2010).
121. Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
122. Neph, S. et al. BEDOPS: high-performance genomic feature operations. Bioinformatics 28, 1919–1920 (2012).
123. Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
124. Rambaut, A. FigTree, version 1.4; http://tree.bio.ed.ac.uk/software/figtree/ (2012).
125. Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
126. GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
127. Gardiner-Garden, M. & Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 196, 261–282 (1987).
128. Zhou, W., Liang, G., Molloy, P. L. & Jones, P. A. DNA methylation enables transposable element-driven genome expansion. Proc. Natl Acad. Sci. USA 117, 19359–19366 (2020).
129. Hartwick, N. T. & Michael, T. P. OrthoBrowser: gene family analysis and visualization. Bioinformatics Adv. 5, vbaf009 (2025).
130. Adami, C. Information theory in molecular biology. Phys. Life Rev. 1, 3–22 (2004).
131. Lovell, J. T. et al. GENESPACE tracks regions of interest and gene copy number variation across multiple genomes. eLife 11, e78526 (2022).
132. R Core Team. R: A Language and Environment for Statistical Computing. http://www. R-project.org/ (R Foundation for Statistical Computing, 2013).
133. Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
134. Li, H. & Ralph, P. Local PCA shows how the effect of population structure differs along the genome. Genetics 211, 289–304 (2019).
135. Calle García, J. et al. PRGdb 4.0: an updated database dedicated to genes involved in plant disease resistance process. Nucleic Acids Res. 50, D1483–D1490 (2022).
136. Mihalyov, P. D. & Garfinkel, A. R. Discovery and genetic mapping of PM1, a powdery mildew resistance gene in Cannabis sativa L. Front. Agron. https://doi.org/10.3389/ fagro.2021.720215 (2021).
137. Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
138. Rost, B. Twilight zone of protein sequence alignments. Protein Eng. 12, 85–94 (1999).
139. Zhou, H.-C., Shamala, L. F., Yi, X.-K., Yan, Z. & Wei, S. Analysis of terpene synthase family genes in Camellia sinensis with an emphasis on abiotic stress conditions. Sci. Rep. 10, 933 (2020).
140. Punta, M. et al. The Pfam protein families database. Nucleic Acids Res. 40, D290–D301 (2012).
141. Eddy, S. R. Accelerated profile HMM searches. PLoS Comput. Biol. 7, e1002195 (2011).
142. Zager, J. J., Lange, I., Srividya, N., Smith, A. & Lange, B. M. Gene networks underlying cannabinoid and terpenoid accumulation in cannabis. Plant Physiol. 180, 1877–1897 (2019).
143. Jin, H., Song, Z. & Nikolau, B. J. Reverse genetic characterization of two paralogous acetoacetyl CoA thiolase genes in Arabidopsis reveals their importance in plant growth and development. Plant J. 70, 1015–1032 (2012).
144. Booth, J. Terpene and isoprenoid biosynthesis in Cannabis sativa. PhD thesis, Univ. of British Columbia (2020).
145. Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
146. Edgar, R. Usearch. OSTI.gov https://www.osti.gov/biblio/1137186 (2010).
147. Abascal, F., Zardoya, R. & Telford, M. J. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38, W7–W13 (2010).
148. Tamura, K., Stecher, G. & Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
149. Wang, J. & Zhang, Z. GAPIT Version 3: boosting power and accuracy for genomic association and prediction. Genomics Proteomics Bioinformatics 19, 629–640 (2021).
150. Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
151. Prentout, D. et al. An efficient RNA-seq-based segregation analysis identifies the sex chromosomes of Cannabis sativa. Genome Res. 30, 164–172 (2020).
152. Lynch, R. Cannabis_Pangenome. Figshare https://figshare.com/projects/Cannabis_ Pangenome/205555 (2024).
153. Lynch, R. Cannabis pangenome. Figshare https://doi.org/10.25452/figshare.plus.c. 7248427.v1 (2024).
154. Lynch, R. et al. Pangenome metadata and statistics. Figshare https://doi.org/10.6084/ m9.figshare.25869319.v2 (2025).
155. CannabisPangenomeShared. GitHub https://github.com/anthony-aylward/ CannabisPangenomeShared (2024).
156. CannabisPangenomeAnalyses. GitHub https://github.com/padgittl/ CannabisPangenomeAnalyses (2024).
157. Woods, P., Price, N., Matthews, P. & McKay, J. K. Genome-wide polymorphism and genic selection in feral and domesticated lineages of Cannabis sativa. G3 13, jkac209 (2022).
Magyar fordítás és szerkesztési megjegyzés
Magyar fordítás és ábraszöveg-adaptáció. A fordítás az eredeti szerzők, a forrás, a DOI és a licenc feltüntetésével készült; a változtatás a magyar nyelvű fordítás és szerkesztés.
Kapcsolódó tanulmányok
Botanikai-rendszertani modell 2020 · Genomikai-taxonómiai áttekintés 2023